 | Информационная система | |
ГОСУДАРСТВЕННАЯ
СИСТЕМА
САНИТАРНО-ЭПИДЕМИОЛОГИЧЕСКОГО НОРМИРОВАНИЯ
РОССИЙСКОЙ ФЕДЕРАЦИИ
4.1. МЕТОДЫ КОНТРОЛЯ. ХИМИЧЕСКИЕ ФАКТОРЫ
Руководство по методам
контроля
качества
и безопасности
биологически активных добавок к пище
Руководство Р 4.1.1672-03
МИНЗДРАВ РОССИИ
МОСКВА
2004
Руководство по методам контроля качества и безопасности биологически активных добавок к пище. - М.: Федеральный центр Госсанэпиднадзора Минздрава России, 2004.
1. Разработано: ГУ НИИ питания РАМН (руководитель В.А. Тутельян, ответственный исполнитель К.И. Эллер, Ю.П. Алешко-Ожевский, Т.В. Аристархова, В.Г. Байков, Н.А. Бекетова, В.В. Бессонов, С.В. Волкович, Л.Ш. Воробьева, О.А. Вржезинская, М.Г. Гаппаров, Н.А. Голубкина, Г.Ф. Жукова, М.Г. Киселева, Т.В. Киселева, В.М. Коденцова, С.Н. Кулакова, Л.Г. Левин, Ф.А. Медведев, Г.В. Никольская, В.В. Пименова, И.М. Скурихин, О.И. Соловьева, В.Б. Спиричев, Л.А. Харитончик, С.А. Хотимченко); Департаментом Госсанэпиднадзора Минздрава России (А.И. Петухов); Федеральным центром Госсанэпиднадзора Минздрава России (И.В. Брагина); Фармакопейным комитетом Минздрава России (В.Л. Багирова, Е.Л. Ковалева); ВИЛАР РАСХН (С.А. Пинеев).
2. Утверждено и введено в действие Главным государственным санитарным врачом Российской Федерации, Первым заместителем Министра здравоохранения Российской Федерации Г.Г. Онищенко 30 июня 2003 г.
3. Вводится впервые.
СОДЕРЖАНИЕ
УТВЕРЖДАЮ
Главный государственный
санитарный врач Российской Федерации,
Первый заместитель Министра
здравоохранения Российской Федерации
Г. Г. Онищенко
30 июня 2003 г.
Дата введения 30 июня 2003 г.
4.1. МЕТОДЫ КОНТРОЛЯ. ХИМИЧЕСКИЕ ФАКТОРЫ
Руководство по методам контроля
качества и безопасности
биологически активных добавок к пище
Руководство Р 4.1.1672-03
Область применения
Настоящее Руководство по методам контроля качества и безопасности биологически активных добавок к пище (далее - руководство) разработано в соответствии с Федеральными законами «О санитарно-эпидемиологическом благополучии населения» от 30.03.99 № 52-ФЗ (Собрание законодательства Российской Федерации, 1999, № 14, ст. 1650), «О качестве и безопасности пищевых продуктов» от 02.01.00 № 29-ФЗ (Собрание законодательства Российской Федерации, 2000, № 2, ст. 150), постановлением Правительства Российской Федерации от 21.12.00 № 987 «О государственном надзоре и контроле в области обеспечения качества и безопасности пищевых продуктов».
Руководство устанавливает методы контроля ингредиентного состава и показателей качество и безопасности биологически активных добавок к пище (далее - БАД).
Руководство предназначено для юридических лиц и индивидуальных предпринимателей, осуществляющих деятельность в сфере производства и оборота БАД, а также для организаций и учреждений государственной санитарно-эпидемиологической службы Российской Федерации (далее - госсанэпидслужбы России), осуществляющих государственный санитарно-эпидемиологический надзор и контроль за безопасностью и эффективностью БАД в соответствии с СанПиН 2.3.2.1078-01 «Гигиенические требования безопасности и пищевой ценности пищевых продуктов» и другими нормативными документами.
Методы контроля, изложенные в руководстве, применяются на этапах экспертизы и регистрации БАД, при разработке и производстве БАД, их ввозе, хранении, транспортировке и реализации на территории Российской Федерации, при разработке нормативной и технической документации, регламентирующей вопросы обращения БАД.
Глава 1
МЕТОДЫ ОПРЕДЕЛЕНИЯ МАКРОНУТРИЕНТОВ
I. Методы определения азотистых соединений
1. Метод определения общего белка
Метод заключается в определении азота по Кьельдалю с последующим пересчетом на белок. Сущность метода состоит в разложении органического вещества пробы кипящей концентрированной серной кислотой с образованием солей аммония, переведении аммония в аммиак, отгонке его в раствор кислоты, количественном учете аммиака титрометрическим методом и расчете содержания азота в исследуемом материале.
Подготовка к испытанию
Среднюю пробу БАД готовят согласно прописи отбора проб по стандарту определения белка на соответствующий продукт, в случае отсутствия стандарта - по техническим условиям на БАД. Для пересчета содержания белка на сухое вещество (в случае необходимости определения данного показателя) определяют влажность исследуемого БАД или пищевого сырья.
Подготовка реактивов и растворов
Приготовление смешанных катализаторов
Катализатор 1. Смешивают 1 весовую часть сернокислой меди и 30 весовых частей сернокислого калия, тщательно растирают в ступке до получения мелкозернистого порошка.
Катализатор 2. Смешивают 10 весовых частей сернокислой меди, 100 весовых частей сернокислого калия и 2 весовые части селена. Тщательно растирают в ступке до получения мелкозернистого порошка.
При приготовлении катализаторов 1 и 2 допускается заменять сернокислый калий надсернокислым калием в том же количестве.
Катализатор 3. Перекись водорода, 30 %-ный водный раствор.
Приготовление 4 %-ного раствора борной кислоты
40 г борной кислоты растворяют в небольшом количестве теплой дистиллированной воды при нагревании и переносят в колбу вместимостью 1000 см3. После охлаждения доводят объем дистиллированной водой до 1000 см3.
Приготовление 0,05 моль/дм3 (0,1 н) раствора серной кислоты
Используют стандарт-титр серной кислоты. Раствор готовят в соответствии с правилами, приложенными к комплекту.
Допускается приготовление 0,05 моль/дм3 раствора серной кислоты из концентрированной серной кислоты в соответствии с ГОСТ 25791.1-83.
Приготовление смешанного индикатора
Растворяют 0,20 г метилового красного и 0,10 г бромкрезолового зеленого в 100 см3 96 %-ного этилового спирта.
Проведение испытания
Приготовление минерализата
Из усредненной измельченной гомогенной пробы исследуемого БАД для анализа взвешивают на обеззоленном фильтре или в пробирке точную навеску, с погрешностью не более 0,1 %. Содержание азота в анализируемой пробе должно быть не менее 10 мг. Навеску количественно переносят в колбу Къельдаля.
Минерализацию осуществляют одним из двух способов.
Способ 1
Добавляют в колбу Къельдаля 1,5 - 2 г смешанного катализатора 1 или 2. После прибавления катализатора осторожно приливают 10 - 15 см3 концентрированной серной кислоты.
Способ 2
Добавляют в колбу Къельдаля 7 - 10 см3 30 %-ной перекиси водорода в качестве окислителя. После прекращения бурной реакции приливают такое же количество концентрированной серной кислоты.
Колбу покрывают стеклянной воронкой и устанавливают на нагреватель так, чтобы ее ось была наклонена под углом 30 - 45° к вертикали. Вначале коблу нагревают умеренно, чтобы предотвратить бурное пенообразование.
При нагревании навеску время от времени помешивают вращательными движениями колбы. После исчезновения пены нагревание усиливают, пока жидкость не будет доведена до постоянного кипения. При этом следят за тем, чтобы на стенках колбы не оставалось черных несгоревших частиц, смывая их легким встряхиванием содержимого колбы или прибавлением небольшого количества серной кислоты.
После того как жидкость обесцветится (допускается слегка зеленоватый оттенок), нагрев продолжают в течение 30 мин.
После охлаждения к содержимому колбы постепенно приливают, взбалтывая, около 70 см3 дистиллированной воды, охлаждают и приступают к отгонке аммиака.
В бачок-парообразователь через воронку наливают дистиллированную воду (несколько больше половины общего объема бачка) и открывают кран на воронке и зажим на отводящей пар трубке в колбу Къельдаля.
Нагревают воду в бачке на газовой горелке или электрической плитке. Присоединяют пустую колбу Къельдаля к каплеуловителю холодильника и воронке для щелочи и после того, как вода в бачке закипит, закрывают кран воронки бачка-парообразователя. Включают холодильник, подставляют под него пустую коническую колбу и в течение 5 - 10 мин «пропаривают» прибор.
После пропаривания открывают краны воронки бачка-парообразователя и воронки для щелочи и закрывают зажим на отводящей пар трубке в колбу Къельдаля.
Под холодильник подставляют вместо пустой конической колбы коническую колбу с предварительно налитыми в нее из пипетки 20 см3 4 %-ной борной кислоты и 5 капель смешанного индикатора или 25 см3 0,05 моль/дм3 раствора серной кислоты. Колбу подставляют под холодильник так, чтобы его кончик был погружен в раствор кислоты на глубину не менее чем 1 см. Вместо пустой колбы Къельдаля присоединяют колбу с сожженной навеской анализируемой пробы.
Закрывают кран воронки для щелочи, наливают в воронку 33 % раствора щелочи и открывают понемногу кран воронки для щелочи при осторожном покачивании колбы Къельдаля, приливают избыток щелочи, при этом цвет раствора должен резко измениться - от прозрачного до синего или бурого. Открывают зажим на отводящей пар трубке в колбу Къельдаля и закрывают остальные краны, при этом пар будет проходить через жидкость в колбе Къельдаля и увлекать аммиак. В холодильнике пар конденсируется. Раствор аммиака попадает в колбу с 0,1 н. раствором серной кислоты. При нормальном кипении объем раствора в приемной колбе через 20 - 30 мин обычно составляет 150 - 180 см3. Конец отгонки можно установить с помощью красной лакмусовой бумажки. Для этого приемную колбу отставляют от аппарата, предварительно обмыв конец холодильника дистиллированной водой, и подставляют лакмусовую бумажку под стекающие капли дистиллята. Если лакмус не синеет, отгон аммиака закончен. Если лакмус синеет, приемную колбу снова подставляют под холодильник и продолжают отгонку. После окончания отгонки приемную колбу опускают, и конец холодильника обмывают дистиллированной водой в приемную колбы. После этого открывают краны на воронке бачка-парообразователя и воронке для щелочи и закрывают зажим, отводящий пар трубки в колбу Къельдаля. Содержимое приемной колбы титруют 0,1 моль/дм3 раствором гидроокиси натрия до перехода окраски в зеленую.
Необходимо параллельно с определением азота в исследуемой пробе проводить определение азота в реактивах («холостой опыт») для внесения соответствующей поправки в результат анализа. Определение азота в реактивах следует повторять каждый раз после замены партии серной кислоты, катализатора или титрованных растворов. Допускается отгонка аммиака (особенно в случае применения больших колб для сжигания) без использования пара непосредственно нагревом колбы на электрическом нагревателе. Проведение отгонки аммиака и все последующие операции проводятся так же, как и с применением пара.
Способ 3
Определение содержания азота проводят на автоматическом анализаторе типа «Къельдек», фирма Текатор, Швеция в соответствие с инструкцией к прибору.
Обработка результатов
Массовую долю азота (X) в испытуемой пробе в процентах от ее массы при проведении отгонки аммиака в борную кислоту вычисляют по формуле
![]() ,
где
,
где
V1 - объем раствора серной кислоты, израсходованный на титрование испытуемого раствора, см3;
V0 - объем раствора серной кислоты, израсходованный на титрование в контрольном опыте, см3;
K - поправка к титру 0,05 ммоль/дм3 раствора серной кислоты, если он приготовлен не из стандарт-титра;
0,0014 - количество азота, эквивалентное 1 см3 раствора серной кислоты, г;
М - масса навески, г.
Массовую долю азота (X) в испытуемой пробе в процентах от ее массы при отгонке аммиака в серную кислоту вычисляют по формуле:
![]() ,
где
,
где
V0 - объем 0,1 моль/дм3 раствора гидроокиси натрия, израсходованный на титрование 0,05 моль/дм3 серной кислоты в контрольном опыте, см3
V1 - объем 0,1 моль/дм3 раствора гидроокиси натрия, израсходованный на титрование серной кислоты в испытуемом растворе, см3;
K - поправка к титру 0,1 моль/дм3 раствора гидроокиси натрия;
0,0014 - количество азота, эквивалентное 1 см3 0,05 моль/дм3 раствора серной кислоты;
М - масса навески, г.
За окончательный результат испытания принимают среднее арифметическое результатов двух параллельных испытаний. Результаты вычисляют до третьего десятичного знака и округляют до второго десятичного знака.
Расчет содержания общего азота в пробе при использовании автоматического анализатора (способ 3) проводят в соответствии с инструкцией к прибору.
Метрологические характеристики
Допустимое расхождение между двумя параллельными определениями (r) не должно превышать значений, вычисляемых по формуле:
r = 0,03 + 0,02×X1, но не более 0,1 % абсолютного содержания общего азота, где
X1 - среднее арифметическое результатов двух параллельных определений, %.
Допустимое расхождение между результатами испытаний, выполненных в двух разных лабораториях (R) должно превышать значений, вычисляемых по формуле:
R = 0,04 + 0,045×Х2, но не более 0,2 % абсолютного содержания общего азота, где
Х2 - среднее арифметическое результатов двух испытаний, выполненных в двух разных лабораториях, %.
Массовую долю азота в пересчете на сухое вещество продукта (Х3), в процентах, вычисляют по формуле:
![]() ,
где
,
где
X1 - массовая доля азота в испытуемой пробе, %;
W - влажность испытуемой пробы, %.
Массовую долю белка (Y) в процентах вычисляют по формуле:
Y = К´Х1, Х2 или Х3, где
К - коэффициент пересчета азота на белок БАД:
с высоким (более 15 %) содержанием липидов - 6,25;
с умеренным (2 - 15 %) содержанием липидов - 6,38;
с низким содержанием липидов - 5,70.
2. Определение аминокислотного состава
Сущность метода заключается в гидролизе образца до аминокислот и последующем количественном определении образовавшихся аминокислот на аминокислотном анализаторе.
Подготовка к испытанию
В пробе определяют общий белок, содержание липидов и влажность по методикам, описанным в настоящем руководстве.
Для жидких БАД учитывают содержащуюся воду в процессе приготовления гидролизующей смеси таким образом, чтобы концентрация соляной кислоты была 6М. При содержании липидов более 5 % проводят обезжиривание способом, указанным в табл. 1. После обезжиривания остаток подсушивают на воздухе и определяют содержание общего белка. Рассчитывают величину навески образца для гидролиза, исходя из соотношения белка к кислоте, представленных в табл. 1, и при условии содержания белка в пробе не менее 5 мг.
Проведение испытания
Три навески БАД или обезжиренного остатка, подготовленных к гидролизу в соответствии с разделом 1, взятых с точностью 0,0001 г, помещают в стеклянную ампулу с оттянутым концом, заливают расчетным количеством соляной кислоты (в случае жидких БАД берут расчетное количество концентрированной кислоты и доводят до концентрации 6М).
Ампулы запаивают, устанавливают в строго вертикальном положении в металлический патрон или фарфоровый стакан с парафином и помещают в сушильный шкаф с заранее отрегулированной температурой 110±2 °С. Нагревание проводят непрерывно в течение 24, 48 и 72 ч. Затем ампулы охлаждают до комнатной температуры. Необходимость трех временных отрезков гидролиза объясняется различиями в скорости отщепления отдельных аминокислот. На основании результатов последующих анализов содержания аминокислот за каждое время гидролиза строят кривую и методом интерполяции или экстраполяции до нулевого времени находят максимальную величину.
Каждую ампулу вскрывают и сразу приступают к удалению соляной кислоты. Если в гидролизате образовался видимый осадок, его удаляют центрифугированием или фильтрованием с последующим доведением фильтрата в мерной колбе до 25 см3 до точного объема. В случае конечного объема больше 5 см3 и при использовании высокочувствительных приборов для удаления соляной кислоты берут аликвоту.
Удаление соляной кислоты проводят одним из следующих способов:
а) помещают ампулу или пробирку в вакуум-эксикатор над гранулированным гидратом окиси натрия (NaOH) на 12 - 18 ч;
б) на роторном испарителе при температуре не выше 60 °С. Для этого гидролизат количественно переносят в грушевидную колбу, ополаскивая ампулу дистиллированной водой.
Таблица 1
Условия подготовки проб к анализу для БАД, содержащих различные уровни липидов
|
БАД с содержанием липидов |
Способ удаления липидов |
Весовое соотношение белок:соляная кислота 6М |
|
|
1 |
низким, менее 2 % |
не требуется |
1:200 |
|
2 |
высоким, более 15 % |
экстракция 10-кратным количеством диэтилового эфира 3 - 4 раза или смесью этанол-хлороформ (1:2) 10-кратным количеством 2 раза |
1:250 |
|
3 |
умеренным, 2 - 15 % |
не требуется |
1:1000 |
Остаток переносят количественно в мерную колбу с помощью нитратного буфера рН 2,2 или раствора соляной кислоты 0,02 М и доводят до метки. Полученный раствор гидролизата подвергают анализу на аминокислотном анализаторе в соответствии с инструкцией прибору.
В случае, если анализ не может быть проведен немедленно, осадок освобождают от следов соляной кислоты путем добавления к нему дистиллированной воды и повторного испарения на роторном испарителе или в вакуумном эксикаторе. Операцию повторяют до полного исчезновения запаха соляной кислоты.
Хранят образец в морозильной камере или нижней камере холодильника при температуре не выше 5 °С, перед анализом разводят цитратным буфером до необходимой концентрации. Обнаруженный осадок отфильтровывают через плотный фильтр.
Метрологические характеристики
Относительное допустимое расхождение между результатами двух параллельных определений, выполненных в одной лаборатории, по отношению к среднему арифметическому значению (Rr) и относительное допустимое расхождение между результатами испытания, выполненных в двух разных лабораториях, по отношению к среднему арифметическому значению (RR) для основных аминокислот при концентрациях, характерных для трех важнейших групп продуктов, приведены в табл. 2.
Таблица 2
Допустимые относительные внутрилабораторные (Rr) и межлабораторные (RR) расхождения результатов определения содержания аминокислот в основных группах БАД
|
С высоким содержанием белка |
С умеренным содержанием белка |
С низким содержанием белка |
||||
|
Rr, % |
RR, % |
Rr, % |
RR, % |
Rr, % |
RR, % |
|
|
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
Лизин |
11 |
25 |
10 |
25 |
12 |
30 |
|
Гистидин |
19 |
32 |
19 |
37 |
20 |
31 |
|
Аргинин |
11 |
29 |
14 |
26 |
12 |
32 |
|
Аспарагиновая кислота |
11 |
24 |
8 |
19 |
10 |
26 |
|
Треонин |
12 |
26 |
12 |
30 |
10 |
24 |
|
Серин |
14 |
28 |
11 |
32 |
16 |
32 |
|
Глутаминовая кислота |
9 |
19 |
8 |
16 |
10 |
22 |
|
Пролин |
20 |
40 |
17 |
42 |
22 |
41 |
|
Глицин |
13 |
24 |
13 |
24 |
15 |
26 |
|
Аланин |
12 |
30 |
13 |
32 |
21 |
38 |
|
Цистин |
20 |
60 |
17 |
55 |
15 |
45 |
|
Валин |
11 |
28 |
10 |
28 |
20 |
39 |
|
Метионин |
18 |
50 |
11 |
35 |
22 |
50 |
|
Изолейцин |
15 |
39 |
11 |
40 |
11 |
50 |
|
Лейцин |
11 |
26 |
13 |
24 |
14 |
39 |
|
Тирозин |
10 |
27 |
14 |
32 |
20 |
45 |
|
Фенилаланин |
17 |
31 |
11 |
44 |
11 |
40 |
|
Оксипролин |
12 |
40 |
14 |
40 |
10 |
28 |
|
Триптофан |
18 |
60 |
19 |
60 |
22 |
65 |
II. Методы определения липидов
1. Методы определения содержания жира в БАД на растительной и жировой основе
1.1. Гравиметрический метод
Метод основан на извлечении сырого жира из БАД растворителем, последующем удалении растворителя, высушивании и взвешивании извлеченного жира.
Подготовка проб к испытанию
Перед началом определений продукт, отобранный из средней пробы по ГОСТ 26312.1-84, тщательно перемешивают, отбирают навеску массой 50 г и измельчают на мельнице.
Приготовление патрона из фильтровальной бумаги
Для приготовления патрона фильтровальную бумагу обезжиривают следующим образом. Листы фильтровальной бумаги свертывают в трубку и помещают в цилиндр с пришлифованной пробкой так, чтобы вся бумага поместилась в цилиндре. В цилиндр перед помещением бумаги наливают 100 - 200 см3 диэтилового эфира или гексана. После того как растворитель поднимется по бумаге до его верхнего края, цилиндр открывают, бумагу вынимают и дают растворителю испариться, затем ножницами от верхнего края отрезают полоску шириной 4 - 5 см, а остальную часть бумаги используют для приготовления патронов. Вату обезжиривают также в цилиндре. Обезжиренную вату и бумагу хранят в закрытой посуде. Обезжиренный прямоугольный кусок бумаги навертывают на деревянную болванку. По мере навертывания свободный край бумаги подворачивают складками для образования донышка патрона. Бумагу и болванку берут таким образом, чтобы стенки патрона получились двойными, а его диаметр был на 0,5 см меньше диаметра экстрактора. На дно патрона кладут кусочек обезжиренной ваты.
Проведение испытания
В патрон из фильтровальной бумаги отвешивают 1 - 5 г
испытуемой пробы, с погрешностью не более 0,01 г, сверху кладут кусочек
обезжиренной ваты. Приготовленный таким образом патрон помещают в экстрактор
аппарата Сокслета так, чтобы он не был выше верхнего изгиба трубки. Колбу
аппарата Сокслета высушивают при температуре 105±5 °С в течение 2 ч и после
охлаждения взвешивают. Колбу наполняют примерно на ![]() объема гексаном или
диэтиловым эфиром и присоединяют к экстрактору. Пускают воду в холодильник и
колбу с растворителем нагревают на водяной или песочной бане. При этом
растворитель, находящийся в колбе, испаряется и в виде паров проходит через
широкую трубку экстрактора в холодильник, где охлаждается и в виде капель
поступает в экстрактор с патроном. При заполнении экстрактора растворителем до
верхнего изгиба сифонной трубки последний переливается в колбу, унося с собой
жир. В течение 1 ч должно быть 7 - 9 сливов растворителя. Экстракцию ведут 2 ч.
Затем патрон удаляют из экстрактора и отгоняют растворитель из колбы в
экстрактор. После заполнения экстрактора до верхнего изгиба сифонной трубки
чистый растворитель сливают из экстрактора, который затем вновь присоединяют к
аппарату Сокслета, и отгоняют оставшийся в колбе растворитель. По окончании
отгонки растворителя отсоединяют экстрактор, колбу выдерживают на бане до
испарения растворителя. После испарения растворителя колбу помещают в сушильный
шкаф и высушивают при температуре 105 ± 5 °С в течение 60
мин, охлаждают в эксикаторе и взвешивают. Последующее взвешивание проводят
после повторной сушки в течение 30 мин. Высушивание и взвешивание повторяют до тех
пор, пока разность результатов двух последовательных взвешиваний будет не более
0,001 г. Одновременно определяют влажность.
объема гексаном или
диэтиловым эфиром и присоединяют к экстрактору. Пускают воду в холодильник и
колбу с растворителем нагревают на водяной или песочной бане. При этом
растворитель, находящийся в колбе, испаряется и в виде паров проходит через
широкую трубку экстрактора в холодильник, где охлаждается и в виде капель
поступает в экстрактор с патроном. При заполнении экстрактора растворителем до
верхнего изгиба сифонной трубки последний переливается в колбу, унося с собой
жир. В течение 1 ч должно быть 7 - 9 сливов растворителя. Экстракцию ведут 2 ч.
Затем патрон удаляют из экстрактора и отгоняют растворитель из колбы в
экстрактор. После заполнения экстрактора до верхнего изгиба сифонной трубки
чистый растворитель сливают из экстрактора, который затем вновь присоединяют к
аппарату Сокслета, и отгоняют оставшийся в колбе растворитель. По окончании
отгонки растворителя отсоединяют экстрактор, колбу выдерживают на бане до
испарения растворителя. После испарения растворителя колбу помещают в сушильный
шкаф и высушивают при температуре 105 ± 5 °С в течение 60
мин, охлаждают в эксикаторе и взвешивают. Последующее взвешивание проводят
после повторной сушки в течение 30 мин. Высушивание и взвешивание повторяют до тех
пор, пока разность результатов двух последовательных взвешиваний будет не более
0,001 г. Одновременно определяют влажность.
Обработка результатов
Массовую долю сырого жира в процентах, в пересчете на сухое вещество, в испытуемой пробе (X) вычисляют по формуле:
![]() ,
где
,
где
М - масса пробы, г;
m1 - масса пустой колбы, г;
m2 - масса колбы с жиром, г;
W - массовая доля влаги в испытуемой пробе, %.
За окончательный результат определения принимают среднее арифметическое результатов (Х1) двух параллельных определений. Вычисления проводят с точностью до 0,1 %.
Метрологические характеристики
Допустимое расхождение двух параллельных определений (r) не должно превышать значений, рассчитанных по формуле:
r = 0,02 + 0,02×Х1, но не более 0,4 % абсолютного содержания жира, где
X1 - среднее арифметическое результатов двух параллельных определений, %.
Допустимое расхождение между результатами испытаний, выполненных в двух разных лабораториях (R), не должно превышать значений, рассчитанных по формуле:
R = 0,04 + 0,035×Х2, но не более 0,8 % абсолютного содержания жира, где
Х2 - среднее арифметическое результатов анализов, выполненных в двух разных лабораториях, %.
1.2. Определение содержания жира в БАД на зерновой основе
Для определения массовой доли жира в БАД на зерновой основе используют три варианта метода:
а) экстракционный метод с предварительным гидролизом навески;
б) рефрактометрический;
в) бутирометрический.
А. Экстракционный метод с предварительным гидролизом навески
Метод основан на извлечении жира из предварительно гидролизованной навески изделия растворителем и определении количества жира взвешиванием после удаления растворителя из определенного объема полученного раствора.
Проведение испытания
Навеску продукта в 1 - 5 г, взвешенную с погрешностью не более 0,05 г, помещают в плоскодонную колбу вместимостью 300 см3, приливают 100 см3 1,5 % соляной кислоты (или 10 см3 5 % серной кислоты), кипятят в колбе с обратным холодильником на кипящей бане 30 мин. Затем колбу охлаждают водой до комнатной температуры, приливают в колбу 50 см3 хлороформа, плотно закрывают хорошо пригнанной пробкой, энергично взбалтывают в течение 15 мин, затем выливают содержимое в центрифужные пробирки и центрифугируют в течение 2 - 3 мин при 3000 об/мин.
В пробирке образуются три слоя. Верхний водный слой отбирают пипеткой. Пипеткой, снабженной резиновой грушей, отбирают хлороформенный раствор жира и фильтруют его в сухую колбу через небольшой ватный тампон, вложенный в узкую часть воронки, причем кончик пипетки должен при этом касаться ваты.
Фильтрат 20 см3 помещают в предварительно доведенную до постоянной массы и взвешенную на аналитических весах колбу вместимостью 100 см3.
Отбор и фильтрация должны производиться в течение 2 мин.
Хлороформ из колбы отгоняют на горячей водяной бане, пользуясь холодильником. Оставшийся в колбе жир сушат до постоянной массы (обычно 1 - 1,5 ч) при температуре 100 - 105 °С, охлаждают в эксикаторе в течение 20 мин и взвешивают колбу на аналитических весах.
Допускается также следующий способ расслаивания.
После гидролиза в охлажденную колбу добавляют 5 см3 раствора аммиака, 50 см3 хлороформа, затем содержимое колбы взбалтывают в течение 15 мин и оставляют на 1 ч для отстаивания. За это время полностью отделяется и четко виден нижний хлороформный слой.
Если расслаивания не произойдет, добавляют еще 2 - 3 см3 аммиака, следя за тем, чтобы реакция по фенолфталеину оставалась кислой.
После расслаивания отбор, фильтрацию, отгон хлороформного слоя и высушивание жира ведут как описано выше.
Примечания.
1. Отгон и фильтрацию растворителя проводят под вытяжкой.
2. При отсутствии хлороформа допускается применение дихлорэтана, который следует хранить в темных склянках.
Обработка результатов
Массовую долю жира (X) в процентах в пересчете на сухое вещество вычисляют по формуле:
![]() ,
где
,
где
М - масса колбы с высушенным жиром, г;
m1 - масса пустой колбы, г;
50 - объем хлороформа, взятого для растворения жира, см3;
m2 - масса навески исследуемого изделия, г;
20 - объем хлороформного раствора жира, взятого для отгона, см3;
W - массовая доля влаги в испытуемом изделии, %.
За окончательный результат испытания принимают среднее арифметическое (Х1) результатов двух параллельных определений. Вычисления проводят с точностью до 0,1 %.
Метрологические характеристики
Допустимое расхождение двух параллельных определений (r) не должно превышать значений, рассчитанных по формуле:
r = 0,02 + 0,02×X1, но не более 0,4 % абсолютного содержания жира, где
X1 - среднее арифметическое результатов двух параллельных определений, %.
Допустимое расхождение между результатами испытаний, выполненных в двух разных лабораториях (R), не должно превышать значений, рассчитанных по формуле:
R = 0,04 + 0,035×Х2, но не более 0,8 % абсолютного содержания жира, где
Х2 - среднее арифметическое результатов анализов, выполненных в двух разных лабораториях, %.
Б. Рефрактометрический метод
Метод основан на извлечении жира из навески образца ά-бромнафталином или ά-хлорнафталином. Массовую долю жира в образце определяют по разности коэффициентов преломления растворителя и раствора жира в растворителе.
Подготовка к испытанию
Определение коэффициента преломления растворителей. Определяют коэффициент преломления ά-бромнафталина или ά-хлорнафталина при температуре 20 °С, нанося 1 - 2 капли этих растворителей на призму рефрактометра.
Определение плотности растворителей
Плотность растворителей (r, г/см3 при 20 °С) определяют пикнометром и вычисляют по формуле:
![]() ,
где
,
где
Q - водное число пикнометра, г;
М - масса растворителя, г.
Взвешивание проводят с погрешностью не более 0,0002 г. Расхождение между параллельными взвешиваниями должно быть не более 0,0005 г.
Калибровка пипеток. Микропипетки калибруют по растворителю, отмеривая ими соответствующий объем растворителя и взвешивая его в стаканчике с погрешностью не более 0,0002 г. Расхождение между параллельными взвешиваниями должно быть не более 0,0005 г.
Из трех взвешиваний берут среднее арифметическое и вычисляют объем пипетки (V) в см3 по формуле:
![]() ,
где
,
где
М - масса растворителя, соответствующая объему взятой пипетки, г;
r - плотность растворителя при температуре 20 °С.
Проведение испытания
При анализе взвешивают 2 г БАД с точностью до 0,05 г и помещают в фарфоровую ступку. Затем калиброванной пипеткой приливают 4 см3 растворителя. Содержимое ступки энергично растирают в течение 3 мин. Смесь переносят из ступки на маленький складчатый фильтр. Первые 2 - 3 капли фильтрата отбрасывают, а последующий фильтрат в количестве 2 - 3 капель помещают на призму рефрактометра и определяют коэффициент преломления.
При анализе изделий с низкой влажностью перед добавлением песка измельченную навеску смачивают 1 см3 воды. Охладив массу, приливают точно 4 - 6 см3 растворителя и вновь все растирают в течение 3 мин, затем добавляют 2 г безводного углекислого натрия, перемешивают, смесь из ступки переносят на складчатый фильтр и фильтруют в стаканчик.
Из полученного фильтрата наносят 2 - 3 капли на призму рефрактометра и определяют коэффициент преломления.
Определение коэффициента преломления проводят при 20±0,2 °С или любой комнатной температуре.
В последнем случае показатель преломления раствора приводят к температуре 20 °С путем внесения поправки по табл. 3.
Отсчет показателя преломления раствора жира можно также производить при любой комнатной температуре без учета поправки на температуру при условии одновременного определения показателя преломления растворителя при той же температуре.
Обработка результатов
Массовую долю жира (X) в процентах в пересчете на сухое вещество вычисляют по формуле:
![]() ,
где
,
где
Vp - объем растворителя, взятого для извлечения жира, см3;
ж - относительная плотность жира при 20 °С;
Пр - коэффициент преломления растворителя;
Прж - коэффициент преломления раствора жира в растворителе;
Пж - коэффициент преломления жира;
М - масса изделия, г;
W - влажность изделия, %.
За окончательный результат испытания принимают среднее арифметическое (X1) двух параллельных определений.
Метрологические характеристики
Допустимое расхождение двух параллельных определений (r) не должно превышать значений, рассчитанных по формуле:
r = 0,03 + 0,035×X1, но не более 0,6 % абсолютного содержания жира, где
X1 - среднее арифметическое результатов двух параллельных определений, %.
Таблица 3
Температурные поправки при определении показателей преломления раствора жира в ά-бромнафталине
|
Поправка |
Т, °С |
Поправка |
Т, °С |
Поправка |
Т, °С |
Поправка |
Т, °С |
Поправка |
|
|
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
10 |
|
отнять от показателя преломления |
|||||||||
|
15,0 |
0,0022 |
16,0 |
0,0018 |
17,0 |
0,0013 |
18,0 |
0,0009 |
19,0 |
0,0004 |
|
15,1 |
0,0022 |
16,1 |
0,0017 |
17,1 |
0,0013 |
18,1 |
0,0008 |
19,1 |
0,0004 |
|
15,2 |
0,0021 |
16,2 |
0,0017 |
17,2 |
0,0012 |
18,2 |
0,0008 |
19,2 |
0,0004 |
|
15,3 |
0,0021 |
16,3 |
0,0016 |
17,3 |
0,0012 |
18,3 |
0,0007 |
19,3 |
0,0003 |
|
15,4 |
0,0020 |
16,4 |
0,0016 |
17,4 |
0,0011 |
18,4 |
0,0007 |
19,4 |
0,0003 |
|
15,5 |
0,0020 |
16,5 |
0,0016 |
17,5 |
0,0011 |
18,5 |
0,0007 |
19,5 |
0,0002 |
|
15,6 |
0,0019 |
16,6 |
0,0015 |
17,6 |
0,0011 |
18,6 |
0,0006 |
19,6 |
0,0002 |
|
15,7 |
0,0019 |
16,7 |
0,0015 |
17,7 |
0,0010 |
18,7 |
0,0006 |
19,7 |
0,0001 |
|
15,8 |
0,0018 |
16,8 |
0,0014 |
17,8 |
0,0010 |
18,8 |
0,0005 |
19,8 |
0,0001 |
|
15,9 |
0,0018 |
16,9 |
0,0014 |
17,9 |
0,0009 |
18,9 |
0,0005 |
19,9 |
0,0000 |
|
прибавить к показателю преломления |
|||||||||
|
20,0 |
0,0000 |
22,0 |
0,0009 |
24,0 |
0,0018 |
26,0 |
0,0026 |
28,0 |
0,0035 |
|
20,1 |
0,0000 |
22,1 |
0,0009 |
24,1 |
0,0018 |
26,1 |
0,0027 |
28,1 |
0,0036 |
|
20,2 |
0,0001 |
22,2 |
0,0010 |
24,2 |
0,0018 |
26,2 |
0,0027 |
28,2 |
0,0036 |
|
20,3 |
0,0001 |
22,3 |
0,0010 |
24,3 |
0,0019 |
26,3 |
0,0028 |
28,3 |
0,0037 |
|
20,4 |
0,0002 |
22,4 |
0,0011 |
24,4 |
0,0019 |
26,4 |
0,0028 |
28,4 |
0,0037 |
|
20,5 |
0,0002 |
22,5 |
0,0011 |
24,5 |
0,0020 |
26,5 |
0,0029 |
28,5 |
0,0037 |
|
20,6 |
0,0003 |
22,6 |
0,0011 |
24,6 |
0,0020 |
26,6 |
0,0029 |
28,6 |
0,0038 |
|
20,7 |
0,0003 |
22,7 |
0,0012 |
24,7 |
0,0021 |
26,7 |
0,0029 |
28,7 |
0,0038 |
|
20,8 |
0,0004 |
22,8 |
0,0012 |
24,8 |
0,0021 |
26,8 |
0,0030 |
28,8 |
0,0039 |
|
20,9 |
0,0004 |
22,9 |
0,0013 |
24,9 |
0,0022 |
26,9 |
0,0030 |
28,9 |
0,0039 |
|
21,0 |
0,0004 |
23,0 |
0,0013 |
25,0 |
0,0022 |
27,0 |
0,0031 |
29,0 |
0,0040 |
|
21,1 |
0,0004 |
23,1 |
0,0014 |
25,1 |
0,0022 |
27,1 |
0,0031 |
29,1 |
0,0040 |
|
21,2 |
0,0005 |
23,2 |
0,0014 |
25,2 |
0,0023 |
27,2 |
0,0032 |
29,2 |
0,0040 |
|
21,3 |
0,0006 |
23,3 |
0,0015 |
25,3 |
0,0023 |
27,3 |
0,0032 |
29,3 |
0,0041 |
|
21,4 |
0,0006 |
23,4 |
0,0015 |
25,4 |
0,0024 |
27,4 |
0,0033 |
29,4 |
0,0041 |
|
21,5 |
0,0007 |
23,5 |
0,0015 |
25,5 |
0,0024 |
27,5 |
0,0033 |
29,5 |
0,0042 |
|
21,6 |
0,0007 |
23,6 |
0,0016 |
25,6 |
0,0025 |
27,6 |
0,0033 |
29,6 |
0,0042 |
|
21,7 |
0,0007 |
23,7 |
0,0016 |
25,7 |
0,0025 |
27,7 |
0,0034 |
29,7 |
0,0043 |
|
21,8 |
0,0008 |
23,8 |
0,0017 |
25,8 |
0,0026 |
27,8 |
0,0034 |
29,8 |
0,0043 |
|
21,9 |
0,0008 |
23,9 |
0,0017 |
25,9 |
0,0026 |
27,9 |
0,0035 |
29,9 |
0,0044 |
Допустимое расхождение между результатами испытаний, выполненных в двух разных лабораториях (R) не должно превышать значений, рассчитанных по формуле:
R = 0,04 + 0,06×Х2, но не более 1,2 % абсолютного содержания жира, где
Х2 - среднее арифметическое результатов анализов, выполненных в двух разных лабораториях, %.
При вычислении процентного содержания жира пользуются показателями преломления и плотности жиров, указанными в табл. 4.
Таблица 4
Коэффициенты преломления жиров
|
Коэффициент преломления |
Плотность |
|
|
Кунжутное масло |
1,4730 |
0,919 |
|
Подсолнечное масло |
1,4748 |
0,919 |
|
Коровье масло |
1,4605 |
0,920 |
|
Маргарин |
1,4690 |
0,928 |
|
Арахисовое масло |
1,4696 |
0,917 |
|
Горчичное масло |
1,4720 |
0,918 |
Примечания
1. Если в исследуемом образце находится неизвестный жир или имеется смесь жиров, поступают следующим образом: 1 - 5 г измельченного изделия заливают трехкратным количеством растворителя (хлороформа, тетрахлоруглерода и др.), взбалтывают в течение 15 мин, вытяжку фильтруют в колбочку, растворитель полностью отгоняют, остаток подсушивают и определяют коэффициент преломления смеси жиров или неизвестного жира.
2. Для смеси жиров или неизвестного жира плотность принимается равной 0,920.
3. При хорошем растирании навески с растворителем в ступке, когда смесь перенесена на фильтр, разрешается стекающие из воронки капли раствора жира и растворитель наносить на призму рефрактометра, не дожидаясь, пока профильтруется вся смесь.
4. Вся работа с органическими растворителями проводится в вытяжном шкафу или хорошо вентилируемой камере.
В. Бутирометрический метод
Метод основан на растворении исследуемой навески в 60 %-ной серной кислоте и отделении слоя жира в молочном бутирометре центрифугированием в присутствии изоамилового спирта, который образует с серной кислотой изоамилово-серный эфир, уменьшающий величину поверхностного натяжения жировых шариков и способствующий слипанию их в единый жировой слой.
Объем выделившегося жира измеряют в градуированной части бутирометра.
Проведение испытания
Из средней пробы готовых образцов отбирают по две навески массой по 2 г каждая. Навески помещают в фарфоровые стаканчики и заливают 9 см3 60 %-ной серной кислоты.
Стаканчики погружают в водяную баню с температурой воды 80 °С и производят растворение навески в серной кислоте в течение 20 мин при периодическом перемешивании стеклянной палочкой.
После растворения навески темную жидкость переносят в молочные бутирометры, смывая остатки из стаканчика с помощью 10 см3 60 % серной кислоты.
В бутирометры осторожно (чтобы не замочить горлышко) приливают по 1 см3 изоамилового спирта, плотно закрывают резиновыми пробками, плавно перемешивают в течение 3 мин и помещают в гнезда водяной бани с температурой воды 80 °С на 5 мин (пробками вниз).
По истечении 5 мин бутирометры вынимают из водяной бани, размещают в молочной центрифуге (пробками к периферии) и центрифугируют 5 мин. После центрифугирования бутирометры снова помещают на 5 мин в водяную баню с температурой воды 80 °С (пробками вниз), после чего вынимают и отмечают высоту желтого жирового слоя над темной жидкостью по числу малых делений градуированной части бутирометра.
Обработка результатов
Массовую долю жира (X) в процентах в пересчете на сухое вещество вычисляют по формуле:
![]() ,
где
,
где
N - высота жирового слоя в бутирометре по числу малых делений;
0,01133 - количество жира, соответствующее одному малому делению бутирометра, г;
W - влажность, %;
Q - навеска изделия, г.
Для удобства и ускорения расчета можно использовать данные табл. 5.
Для дальнейшего пересчета на сухое вещество умножают массовую долю жира в процентах, найденную по таблице, на 100 и делят на разность (100 - W). За окончательный результат испытания принимают среднее арифметическое (X1) результатов двух параллельных определений.
Таблица 5
Показания бутирометра в зависимости от содержания жира
|
Массовая доля жира, % |
Показания бутирометра |
Массовая доля жира, % |
Показания бутирометра |
Массовая доля жира, % |
Показания бутирометра |
Массовая доля жира, % |
|
|
1 |
0,57 |
11 |
6,23 |
21 |
11,90 |
31 |
17,56 |
|
2 |
1,13 |
12 |
6,80 |
22 |
12,46 |
32 |
18,13 |
|
3 |
1,70 |
13 |
7,36 |
23 |
13,03 |
33 |
18,69 |
|
4 |
2,27 |
14 |
7,93 |
24 |
13,60 |
34 |
19,26 |
|
5 |
2,83 |
15 |
8,50 |
25 |
14,16 |
35 |
19,82 |
|
6 |
3,40 |
16 |
9,06 |
26 |
14,73 |
36 |
20,39 |
|
7 |
3,96 |
17 |
9,63 |
27 |
15,29 |
37 |
20,96 |
|
8 |
4,53 |
18 |
10,19 |
28 |
15,66 |
38 |
21,53 |
|
9 |
5,10 |
19 |
10,76 |
29 |
16,42 |
39 |
22,09 |
|
10 |
5,66 |
20 |
11,33 |
30 |
17,00 |
40 |
22,66 |
Метрологические характеристики
Допустимое расхождение двух параллельных определений (r) не должно превышать значений, рассчитанных по формуле:
r = 0,02 + 0,02×X1, но не более 0,4 % абсолютного содержания жира, где
X1 - среднее арифметическое результатов двух параллельных определений, %.
Допустимое расхождение между результатами испытаний, выполненных в двух разных лабораториях (R), не должно превышать значений, рассчитанных по формуле:
R = 0,04 + 0,04×X2, но не более 0,8 % абсолютного содержания жира, где
Х2 - среднее арифметическое результатов анализов, выполненных в двух разных лабораториях, %.
1.3. Определение массовой доли жира в БАД с высоким содержанием жира методом экстракции в аппарате Сокслета
Метод предназначен для определения массовой доли жира в БАД с высоким содержанием жира при проведении исследований и арбитражного анализа.
Диапазон измеряемых концентраций от 40 до 85 %.
Метод основан на экстракции растворителем в аппарате Сокслета.
Подготовка к испытанию
Гигроскопическую вату и фильтровальную бумагу помещают в аппарат Сокслета и экстрагируют этиловым эфиром в течение 2 - 3 ч.
Подготовка экстракционных патронов
Патроны для экстракционных насадок типа НЭТ готовят как указано выше.
Подготовка БАД к экстракции
В фарфоровую ступку взвешивают по разности 5 г БАД с записью значения массы до четвертого десятичного знака. Смешивают с 15 г прокаленного сульфата натрия и переносят в подготовленный экстракционный патрон, после чего ступку и шпатель с помощью пинцета протирают несколько раз ватой, сначала сухой, а затем смоченной этиловым эфиром. Всю вату помещают в тот же патрон. Сверху кладут небольшой слой ваты, затем края патрона завертывают.
Экстракция жира
Патрон с навеской БАД помещают в экстрактор. К
экстрактору присоединяют чистую колбу, предварительно высушенную в течение 1 ч
при 100 - 105 °С и взвешенную после охлаждения. Через холодильник при помощи
воронки наливают в экстрактор этиловый эфир так, чтобы патрон в экстракторе был
полностью покрыт слоем эфира. В колбу наливают также эфир на ![]() ее объема.
ее объема.
Колбу собранного аппарата нагревают на закрытой водяной бане, обеспечивающей равномерное, не слишком сильное кипение эфира. Через 3 ч проверяют полноту экстракции. Для этого, охладив колбу, отсоединяют ее на мгновение от остальной части прибора и 1 - 2 капли эфира с нижнего конца сифона экстрактора наносят на чистое часовое стекло или кусочек фильтровальной бумаги. Экстракцию считают законченной, если после испарения эфира не остается масляных пятен на стекле или бумаге.
По окончании аппарат разбирают, вынимают патрон из экстрактора, последний присоединяют снова к колбе и отгоняют растворитель на водяной бане. Колбу сушат с жиром в сушильном шкафу в течение 1 ч при температуре 100 - 105 °С. Колбу взвешивают после охлаждения ее в эксикаторе. Последующие взвешивания производят через каждые 15 мин сушки. Масса считается постоянной, если отличается от предыдущей не более чем на 0,0004 г.
Расчет результата определения
Массовую долю жира в БАД в % (Х) вычисляют по формуле:
![]() ,
где
,
где
М1 - масса колбы с высушенным жиром, г;
М2 - масса пустой колбы, г;
М - навеска БАД, г.
Конечным результатом считают среднее арифметическое двух параллельных определений (Х1) отдельных проб БАД. Вычисления проводят до второго десятичного знака.
Метрологические характеристики
Допустимое расхождение двух параллельных определений (r) не должно превышать значений, рассчитанных по формуле:
r = 0,02 + 0,01×X1, но не более 0,4 % абсолютного содержания жира, где
X1 - среднее арифметическое результатов двух параллельных определений, %.
Допустимое расхождение между результатами испытаний, выполненных в двух разных лабораториях (R), не должно превышать значений, рассчитанных по формуле:
R = 0,04 + 0,02×X2, но не более 0,8 % абсолютного содержания жира, где
Х2 - среднее арифметическое результатов анализов, выполненных в двух разных лабораториях, %.
2. Методы определения жирнокислотного состава
Метод основан на разделении метиловых эфиров жирных кислот, полученных из липидов БАД, с помощью газожидкостной хроматографии.
Выделение липидов из БАД
Липиды выделяют экстракционным методом, предложенным для данной группы БАД, исключающим термическое воздействие на объект и изложенным в вышеприведенных разделах.
Получение метиловых эфиров жирных кислот
Метанолиз в щелочной среде нейтральных масел и жиров проводят по ГОСТ 30418-96 «Масла растительные. Метод определения жирнокислотного состава». Получение метиловых эфиров жирных кислот может быть проведено метанолизом в кислой среде или переэтерификацией липидов метенолом в среде хлористого водорода по ГОСТ Р 51486-99 «Масла растительные и жиры животные. Получение метиловых эфиров жирных кислот».
Для определения абсолютных количеств жирных кислот проводится анализ с применением внутреннего стандарта. В качестве внутреннего стандарта используют маргариновую кислоту или дибутилфталат.
Анализ метиловых эфиров жирных кислот методом ГЖХ
Метод основан на использовании газо-жидкостной хроматографии (ГЖХ) метиловых эфиров жирных кислот по ГОСТ Р 51483-99 «Масла растительные и жиры животные. Определение методом газовой хроматографии массовой доли метиловых эфиров индивидуальных жирных кислот к их сумме». Результаты выражают в массовых долях каждого индивидуального компонента БАД.
Эталонный стандарт - смесь метиловых эфиров известного состава, желательно, аналогичного составу анализируемого вещества.
Идентификацию пиков метиловых эфиров жирных кислот проводят по относительным приведенным временам удерживания или эквивалентным длинам цепи (ЭДЦ).
В качестве неподвижных жидких фаз используют полиэфиры типа полиэтиленгликольадипат, полипропиленгликольадипат, диэтиленгликольсукцинат, цианосиликоны (SP-100, FFAP, силары OV-275, Sil-88) или другие жидкости, дающие необходимое хроматографическое разделение.
Комментарий к условиям анализа
Температура термостата колонок:
а) при анализе смесей метиловых эфиров жирных кислот с длиной цепи более 14 углеродных атомов 170 - 190 °С (изотермический режим). Условия подбирают таким образом, чтобы метилолеат выходил из колонки за 15 мин;
б) при анализе смесей метиловых эфиров, содержащих низкомолекулярные компоненты, температуру термостата программируют в пределах от 120 до 195 °С со скоростью 6 - 8 °С в 1 мин (при наличии метилового эфира масляной кислоты начальная температура программирования 70 °С).
Величина вводимой пробы - около 1 - 2 мкл раствора метиловых эфиров в гексане для набивных колонок и 0,2 - 0,3 мкл - для капиллярных.
В случае необходимости рекомендуется производить анализ на двух зафиксированных фазах с различными полярностями с целью проверки отсутствия скрытых пиков (для рыбьего жира или в случае одновременного присутствия С18:3, С20:0, С20:1).
Обработка результатов
Качественный анализ
Анализируют эталонную смесь метиловых эфиров. Строят график зависимости логарифма времени удерживания от числа углеродных атомов в цепи. Получают ряд параллельных прямых для насыщенных, моно-, ди- и т.д. ненасыщенных метиловых эфиров кислот или находят величины эквивалентной длины цепи ЭДЦ для ненасыщенных и разветвленных жирных кислот по формуле:
![]() ,
где
,
где
n - число атомов углерода в насыщенной кислоте нормального строения, находящейся на хроматограмме перед неизвестным компонентом;
lgVn - логарифм времени удерживания кислоты с n-числом атомов углерода;
lgVn+1 - логарифм времени удерживания кислоты с n+1-числом атомов углерода;
lgVx - логарифм времени удерживания неизвестного компонента.
Идентифицируют пики на хроматограмме анализируемой смеси по полученному графику или по величинам ЭДЦ. Следует избегать условий анализа, при которых происходит наложение пиков метиловых эфиров различных кислот (например, метиллинолената и метиларахината).
Количественный анализ и способы расчета
За исключением специальных анализов расчет проводят методом внутренней нормализации, когда все компоненты смеси представлены на хроматограмме и площадь всех пиков компонентов составляет 100 %. При наличии интегратора пользуются полученными с его помощью цифрами. При отсутствии интегратора площади пиков измеряют, умножая высоту пика на его ширину (или ширину пика на половине его высоты), и учитывая переключения чувствительности усилителя, используемые во время записи хроматограммы.
Расчет без использования поправочных коэффициентов
Расчет массовой доли i-го компонента в % проводят по формуле:
![]() ,
где
,
где
С - массовая доля i-го компонента;
Si - площадь пика i-го компонента;
![]() - сумма всех площадей пиков.
- сумма всех площадей пиков.
За результат анализа принимают среднее арифметическое (X1) из двух параллельных измерений.
Расчет с использованием поправочных коэффициентов
В случае, когда в анализируемой смеси присутствуют кислоты с 12 и менее углеродными атомами в цепи, расчет ведется с применением поправочных коэффициентов.
Поправочные коэффициенты (Кi) рассчитывают по хроматограммам эталонных смесей известного состава, полученным в условиях, применяемых для анализа образца по формуле:
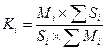 ,
где
,
где
Mi - масса i-го компонента в эталонной смеси, г;
![]() - масса эталонной смеси, г.
- масса эталонной смеси, г.
Часто используют относительные поправочные
коэффициенты (![]() ) по
отношению к поправочному коэффициенту метилпальмитата (
) по
отношению к поправочному коэффициенту метилпальмитата (![]() ):
):
![]() .
.
Массовую долю i-го компонента анализируемой смеси определяют по формуле:
![]() .
.
За результат анализа принимают среднее арифметическое из двух параллельных измерений с записью до первого десятичного знака.
Расчет с применением внутреннего стандарта
В случаях, когда не все компоненты в анализируемой смеси элюируются из колонки или присутствуют очень летучие компоненты, используют для расчета метод внутреннего стандарта.
В качестве внутреннего стандарта применяют метиловые эфиры кислот, отсутствующие в анализируемой смеси. При анализе проб, содержащих масляную кислоту, в качестве внутреннего стандарта используют валериановую кислоту, в остальных случаях - пентадекановую или маргариновую кислоту. Массовую долю i-го компонента пробы определяют по формуле:
![]() ,
где
,
где
т - содержание липидов в продукте, мг в 100 г;
Ms - масса внутреннего стандарта, мг;
М - масса образца, мг;
Ks - поправочный коэффициент внутреннего стандарта;
Ss - площадь пика внутреннего стандарта.
Метрологические характеристики
Допустимые расхождения содержания жирных кислот в продукте рассчитывают (в %) по следующим формулам:
r = 0,225 + 0,04 Х1, но не более 1 % абсолютного содержания кислоты,
R = 0,310 + 0,075×Х2, но не более 3 % абсолютного содержания кислоты.
3. Методы определения стеринов
3.1. Колориметрический метод определения содержания стеринов после омыления проб
Метод основан на колориметрической реакции стеринов, извлекаемых диэтиловым эфиром из омыленных проб растительных масел, с уксусным ангидридом в присутствии концентрированной серной кислоты.
Условия проведения анализа
Экстракцию неомыленных веществ следует проводить по возможности быстро, предохраняя пробы от попадания на них прямого солнечного света.
Отгонку диэтилового эфира из проб следует проводить под вакуумом водоструйного насоса при комнатной температуре.
Подготовка к проведению анализа
Приготовление и очистка реактивов
Безводный сернокислый натрий прокаливают в течение 1 - 1,5 ч при температуре 110 °С. Диэтиловый эфир обрабатывают марганцовокислым калием (5 г на л) и гидратом окиси калия (10 г на 1 дм3) в течение суток, а затем перегоняют. Этиловый спирт ректификованный технический освобождают от альдегидов. Начальную и конечную порции отгона отбрасывают. Хлороформ сушат в течение суток под хлористым кальцием и перегоняют.
Построение градуировочного графика
Градуировочный график строят на основании результатов анализа проб с известным содержанием чистого b-ситостерина.
В мерную колбу вместимостью 100 см3 отвешивают 0,15 - 0,20 г b-ситостерина (с записью результата до 4-го знака после запятой). Навеску растворяют в 100 см3 хлороформа. Из полученного раствора готовят серию стандартных растворов с содержанием b-ситостерина 0,02; 0,04; 0,06; 0,08; 0,10; 0,12; 0,14; 0,16; 0,18; 0,20 г/дм3. Из каждого раствора отбирают пипеткой 3 см3, добавляют 2 см3 уксусного ангидрида и 6 капель серной кислоты. Через 10 мин после добавления кислоты измеряют оптическую плотность раствора на спектрофотометре при 690 нм. В качестве контроля служит раствор, состоящий из 3 см3 хлороформа, 2 см3 уксусного ангидрида и 6 капель концентрированной серной кислоты.
Градуировочный график строят в координатах: оптическая плотность (D) - концентрация стандартных растворов b-ситостерина. Градуировочный график строят для каждого спектрофотометра и проверяют при смене партий путем определения оптической плотности стандартных растворов двух разных концентраций.
Проведение анализа
Испытуемый образец (1 - 3 г) взвешивают в колбе вместимостью 100 см3 (с записью результата взвешивания до 4-го знака после запятой), добавляют 0,1 - 0,3 г аскорбиновой кислоты и 10 - 30 см3 свежеприготовленного 2 н. спиртового раствора КОН. Смесь нагревают с обратным холодильником на кипящей водяной бане в течение 30 мин.
Содержимое колбы охлаждают и количественно переносят в делительную воронку с 30 - 90 см3 дистиллированной воды. Неомыляемые вещества экстрагируют диэтиловым эфиром 3 - 4 раза порциями по 20 - 60 см3. Объединенный эфирный экстракт промывают в делительной воронке дистиллированной водой до нейтральной реакции промывной воды по фенолфталеину. Промытую эфирную вытяжку помещают в коническую колбу, засыпают 5 - 10 г безводного сульфата натрия и оставляют в темном месте на 30 мин, периодически взбалтывая. Затем содержимое колбы фильтруют через бумажный фильтр в другую колбу, фильтр ополаскивают эфиром. Эфир отгоняют под вакуумом при температуре не выше 25 - 30 °С.
Остаток в колбе после отгонки эфира растворяют в зависимости от навески исследуемого образца в 1 - 3 см3 бензола. Затем производят разделение компонентов смеси неомыляемых веществ методом тонкослойной хроматографии. Для этого на пластинку «Силуфол» наносят 50 - 150 мкл бензольного раствора неомыляемых веществ в виде полоски, отстоящей на 2 см от нижнего и боковых краев пластинки.
На одном уровне с пробой на расстоянии 1 см от краев пластинки наносят по капле раствор b-ситостерина. Пластинку помещают вертикально в хроматографическую камеру, в которую заранее наливают смесь диэтилового и петролейного эфиров, взятых в соотношении 1:1. Количество растворителя зависит от размеров хроматографической камеры и регулируется высотой его слоя, равной 1 см.
Развитие хроматограммы продолжается до тех пор, пока фронт растворителя не поднимается на 10 - 12 см. Обычно это занимает 10 - 12 мин. Затем пластинку вынимают из камеры и подсушивают на воздухе до исчезновения запаха эфира. Края хроматограммы шириной 2 см опрыскивают 5 %-ным спиртовым раствором фосфорно-молибденовой кислоты, после чего пластинки помещают на 1 - 5 мин (до проявления) в термостат с температурой около 90 °С. На уровне окрашенного пятна свидетеля соскребают слой адсорбента. Адсорбент элюируют хлороформом 6 - 8 раз порциями по 1 см3. После каждого элюирования адсорбент отделяют центрифугированием или фильтрацией. Объединенный элюат собирают в градуированную пробирку и упаривают до объема 3 см3. К хлороформному раствору стеролов приливают 2 см3 уксусного ангидрида и 6 капель концентрированной серной кислоты (по капле). Через 10 мин измеряют оптическую плотность раствора на спектрофотометре при 690 нм.
Контролем служит раствор, состоящий из 3 см3 хлороформа, 2 см3 уксусного ангидрида и 6 капель концентрированной серной кислоты.
Обработка результатов
Массовую долю стеринов в пробе (X, %) рассчитывают по формуле:
![]() ,
где
,
где
V - объем хлороформного раствора, взятого для проведения колориметрической реакции, см3;
V1 - объем бензольного раствора неомыляемых веществ, см3;
V2 - объем бензольного раствора неомыляемых веществ, нанесенный на пластинку, см3;
С - концентрация стеролов, определяемая по градуировочному графику, г/дм3;
М - масса образца, г.
За окончательный результат испытания принимают среднее арифметическое (X) результатов двух параллельных определений.
Метрологические характеристики
Относительное допустимое расхождение между результатами двух параллельных определений, выполненных в одной лаборатории, по отношению к среднему арифметическому значению (Rr) и относительное допустимое расхождение между результатами испытаний, выполненных в двух разных лабораториях, по отношению к среднему арифметическому значению (RR) приведены в табл. 6.
Таблица 6
Относительные допустимые внутрилабораторные (Rr) и межлабораторные (RR) расхождения результатов определения
|
Rr, % |
RR, % |
|
|
Менее 0,5 |
20 |
40 |
|
0,5 - 1,0 |
15 |
30 |
|
Более 1,0 |
10 |
20 |
3.2. Определение состава стеринов методом ГЖХ
Метод основан на прямом газохроматографическом анализе стеринов, выделенных из липидов путем тонкослойной хроматографии фракции неомыляемых веществ.
Условия проведения анализа
Выделение липидов проводят одним из унифицированных методов, предложенным для данного продукта и исключающим разрушение стеринов.
Экстракцию неомыляемых веществ следует проводить по возможности быстро, предохраняя пробы от попадания на них прямого солнечного света.
Отгонку диэтилового эфира из проб следует проводить под вакуумом водоструйного насоса при комнатной температуре.
Подготовка к проведению анализа
Подготовку проводят по п. «Колориметрический метод определения содержания стеринов».
Проведение анализа
Выделение стеролов из липидов
Омыление липидов, экстракцию неомыляемых веществ, выделение стеринов методом тонкослойной хроматографии проводят по п. «Колориметрический метод определения содержания стеринов».
Газохроматографический анализ
Аппаратура, используемая для проведения анализа, выбор оптимальных условий для разделения стеринов, определение эффективности хроматографических колонок изложены в п. выше.
Устанавливают следующие условия анализа на хроматографе: стеклянная или стальная колонка длиной 1,5 - 2,0 м и внутренним диаметром 3 - 4 мм, заполненная силанизированным целитом 545 (60 - 100 меш), обработанным 3 % полиметилсилоксаном SE-30 или аналогичной неподвижной фазой. Скорость потока газа-носителя 60 - 100 см3/мин. Температура термостата колонки 250 - 270 °С, инжектора 300 °С, детекторов 280 °С.
Вводят в хроматограф около 3 мкл каждого компонента.
Обработка результатов
Рассчитывают площади пиков каждого компонента (Si) по формуле:
Si = Ki´Di´Hi, где
Di - ширина i-го пика на половине его высоты, мм;
Hi - высота i-го пика, мм;
Ki - поправочный коэффициент.
Для получения поправочных коэффициентов составляют 3 - 4 стандартные смеси стеролов, входящих в исследуемые образцы, и холестерола. По хроматограммам стандартных смесей, полученных в идентичных условиях, определяют поправочные коэффициенты по отношению к холестеролу, коэффициент которого принят за единицу, по формуле:
![]() ,
где
,
где
mi и Мст - масса i-го компонента и стандарта (холестерина);
Si и Sст - площади их пиков соответственно.
Молярную долю каждого стерина (Сi) рассчитывают методом нормализации по формуле, принимая сумму площадей всех типов за 100 %:
![]() ,
где
,
где
![]() - площадь пика i-го компонента;
- площадь пика i-го компонента;
![]() - сумма
площадей пиков стеринов.
- сумма
площадей пиков стеринов.
За окончательный результат испытания принимают среднее арифметическое (X) результатов двух параллельных определений.
Метрологические характеристики
Относительное допустимое расхождение между результатами двух параллельных определений, выполненных в одной лаборатории по отношению к среднему арифметическому значению (Rr), и относительное допустимое расхождение между результатами испытаний, выполненное в двух разных лабораториях по отношению к среднему арифметическому значению (RR) приведены в табл. 7.
Таблица 7
Относительные допустимые внутрилабораторные (Rr) и межлабораторные (RR) расхождения результатов определения
|
Rr, % |
RR, % |
|
|
Менее 0,5 |
20 |
40 |
|
0,5 - 1,0 |
15 |
30 |
|
Более 1,0 |
10 |
20 |
3.3. Метод хромато-масс-спектрометрии в анализе стеринов
Метод основан на прямом хромато-масс-спектрометрическом анализе стеринов, выделенных из липидов путем тонкослойной хроматографии фракции неомыляемых веществ.
Условия проведения анализа
Выделение липидов проводят одним из унифицированных методов, предложенным для данного БАД и исключающим разрушение стеринов.
Экстракцию неомыляемых веществ следует проводить по возможности быстро, предохраняя пробы от попадания на них прямого солнечного света, отгонку растворителя следует проводить под вакуумом на ротационном испарителе при комнатной температуре.
Подготовку к проведению анализа и выделение стеринов из липидов проводят по п. «Колориметрический метод определения содержания стеринов».
Условия анализа: хромато-масс-спектрометр с автоматической системой обработки данных, энергия ионизирующих электронов 70 eV; диапазон массовых чисел 39 - 500 а.е.м.; температура источника ионов 120 °С; хроматографическое разделение на капиллярной колонке 47 м´0,3 мм с химически связанной фазой SE-30, начальная температура колонки 50 °С - 2 мин, 240 °С - 8 мин. с последующим программированием со скоростью 4 °С/мин до 300 °С и изотермой (30 мин), температура испарителя 280 °С, переходных линий 275 °С. Скорость газа-носителя гелия 1,3 см3/мин, в момент ввода пробы сброс перекрыт на 30 с, далее деление потока 1:60.
Структуру компонентов определяют путем сравнения полученных масс-спектров с имеющейся базой данных, а также по молекулярным и характеристическим осколочным ионам, выбор которых определяется эмпирическими корреляциями между масс-спектром и структурой изучаемых стеринов.
Таблица 8
Состав фракций стеринов, % отн., выделенных из ряда БАД на растительной основе и на основе морепродуктов, полученный с помощью метода хромато-масс-спектрометрии
|
Стерины |
М.м. |
соя |
авокадо |
апельсин |
кокос |
амарант (ширица) |
рапс |
кальмар |
|
D-5, 7, 22, 24-эргостатетраенол |
394 |
0,7 |
|
|
|
|
|
|
|
4a-метил-холеста-5, 7, 22-триенол |
396 |
0,4 |
|
|
|
|
|
|
|
холестерин |
386 |
0,1 |
0,4 |
0,8 |
0,7 |
2,3 |
0,7 |
96,8 |
|
десмостерин |
384 |
|
|
|
|
|
|
1,6 |
|
брассикастерин |
398 |
|
|
|
|
|
10,3 |
|
|
эргостерин |
396 |
|
|
0,3 |
|
|
|
|
|
24-метиленхолестерин |
398 |
|
|
0,7 |
|
|
|
1,0 |
|
кампестерин |
400 |
17,4 |
4,0 |
4,0 |
5,8 |
- |
29,4 |
|
|
дигидрокампестерин |
402 |
1,0 |
|
|
|
|
2,0 |
|
|
лофенол |
400 |
|
|
|
|
|
|
0,4 |
|
стигмастерин |
412 |
16,1 |
0,7 |
6,4 |
9,5 |
|
|
|
|
D-7-кампестерин |
400 |
|
|
|
|
4,3 |
|
|
|
обтусифолиол |
426 |
|
|
|
0,6 |
|
|
|
|
клионастерин |
414 |
|
|
|
|
|
|
0,2 |
|
циклоартанол |
428 |
|
|
|
0,7 |
|
|
|
|
b-ситостерин |
414 |
47,5 |
89,9 |
83,5 |
44,2 |
- |
52,9 |
|
|
дигироситостерин |
416 |
3,0 |
|
|
|
|
|
|
|
b-амирин |
426 |
1,4 |
|
|
|
|
|
|
|
D-5-авенастерин |
412 |
2,4 |
3,8 |
2,2 |
10,2 |
|
4,7 |
|
|
a-амирин |
426 |
1,4 |
|
|
|
|
|
|
|
циклоэфкаленол |
426 |
1,1 |
|
|
2,3 |
|
|
|
|
циклоартенол |
426 |
2,4 |
0,1 |
|
16,2 |
1,0 |
|
|
|
D-7-стигмастерин |
412 |
2,6 |
0,4 |
0,6 |
4,4 |
60,6 |
|
|
|
D-7-авенастерин |
412 |
1,4 |
0,6 |
1,0 |
|
|
|
|
|
24-метиленциклоартанол |
440 |
|
0,1 |
|
4,1 |
|
|
|
|
D-7-ситостерин |
414 |
|
|
|
|
31,8 |
|
|
|
цитростадиенол |
426 |
2,4 |
|
0,5 |
1,3 |
|
|
|
4. Метод определения фосфолипидов (определение суммарного фосфора)
Метод основан на способности фосфора, соединяясь с молибденом аммония образовывать фосфорномолибденовую кислоту, которая восстанавливается амидоловым реагентом и дает голубое окрашивание.
Специфические приборы и реактивы
Спектрофотометр.
Молибдат аммония, 8,4 %-ный.
Амидоловый реагент, 1 %-ный, свежеприготовленный.
Спиртовой раствор фосфора, 1,097 г KН2PО4 растворяют в 250 см3 воды, этот запасной раствор разбавляют водой и получают рабочий раствор, содержащий 10 мкг фосфора в 1 см3.
Подготовка проб
Пипеткой отбирают аликвотную часть раствора липидов (содержащую 10 - 100 мкг фосфора, но не более 2 мг липидов) и переносят ее в толстостенную пирексовую пробирку с делениями, соответствующими 12,5 и 25 см3. Растворитель упаривают досуха в токе воздуха при 350 °С, к остатку прибавляют 2,0 хлорной кислоты, немного стеклянных шариков и нагревают пробирку на электрической плитке или газовой горелке до тех пор, пока раствор не станет прозрачным и бесцветным. Охлажденный раствор разбавляют водой по 12,5 см3, тщательно перемешивают, добавляют 1,0 см3 раствора молибдата аммония, перемешивают и оставляют на 20 мин до появления голубой окраски.
Проведение испытания
Окрашенный раствор разбавляют водой по 25 см3, перемешивают. На спектрофотометре типа СФ-46 в парных кюветах 1 см определяют поглощение при 680 нм полученного раствора и р-ра холостого опыта.
Обработка результатов
Расчет производят по калибровочной кривой, построенной по стандартному раствору KН2PО4. Используют аликвотные пробы стандартного раствора, содержащие от 30 до 60 мкг фосфора. Закон Бера соблюдается в интервале 10 - 100 мкг фосфора.
III. Методы определения углеводов
1. Определение содержания крахмала с помощью поляриметрического метода
Метод определения массовой доли крахмала в БАД на зерновой основе распространяется на продукты с массовой долей крахмала выше 10 %. Содержание крахмала определяют поляриметрическим методом путем его гидролиза раствором соляной кислоты.
Специфические аппаратура, материалы и реактивы
Сахариметр или поляриметр.
Мельница лабораторная, обеспечивающая требуемую крупность размола.
Сито из проволочной сетки № 08 по ТУ 14-4-1374-86.
Соляная кислота по ГОСТу 3118-77, раствор массовой концентрацией 11,24 г/дм3, для анализа картофеля - 3,77 г/дм3.
Калий железистосинеродистый по ГОСТу 4207-75, раствор массовой концентрацией 150 г/дм3.
Цинка сульфат по ГОСТу 3765-78, раствор массовой концентрацией 150 г/дм3.
Аммония молибдат по ГОСТу 3765-78, раствор массовой концентрацией 100 г/дм3.
Натрия молибдат по ГОСТу 10931-74, раствор массовой концентрацией 150 г/дм3.
Фосфорно-вольфрамовая кислота, раствор массовой концентрацией 40 г/дм3.
Подготовка к испытанию
Пробы, влажность которых превышает 17 %, предварительно подсушивают на воздухе или в одном из следующих устройств: сушильном шкафу, термостате, лабораторном сушильном аппарате при температуре воздуха не более 50 °С. Пробу тщательно перемешивают, измельчают до такой степени, чтобы весь размолотый материал прошел при просеивании через сито из проволочной сетки № 8.
Одновременно со взятием навесок для анализа берут навески для определения влажности. Определение влажности осуществляется по ГОСТам, принятым в соответствующей отрасли.
Проведение испытания
Из аналитической пробы берут навеску массой 5 г с погрешностью не более 0,01 г, помещают в 100 см3 центрифужный стакан, доливают 18 см3 10 % раствора этанола (при определении массовой доли крахмала в отрубях - 28 см3 раствора этанола) и перемешивают стеклянной палочкой. Стеклянную палочку ополаскивают 2 см3 раствора этанола. Закрывают центрифужный стакан резиновой пробкой и вручную сильно встряхивают в течение 2 мин.
После встряхивания стенки центрифужного стакана и резиновую пробку ополаскивают 25 см3 этанола. Затем в течение 20 мин пробу центрифугируют при 4000 об/мин, после чего прозрачный центрифугат сливают. К остатку постепенно добавляют 20 см3 соляной кислоты, перемешивают стеклянной палочкой до образования суспензии и переносят в мерную колбу на 100 см3. Прилипшие к стенкам центрифужного стакана и к стеклянной палочке остатки пробы многократно ополаскивают раствором соляной кислоты массовой концентрацией 11,24 г/дм3 в мерную колбу; общее количество раствора соляной кислоты составляет 50 см3.
Мерную колбу при постоянном встряхивании погружают в кипящую водяную баню. Из-за погружения мерной колбы не должен нарушаться процесс кипения водяной бани: с помощью специальных уплотнительных колец ее следует держать по возможности закрытой. По секундомеру встряхивают мерную колбу в течение 3 мин, при этом колбу из водяной бани не поднимают. После этого выдерживают колбу без встряхивания для всех крахмалосодержащих БАД 12 мин.
По истечении в общей сложности 15 мин для всех крахмалосодержащих БАД, колбу вынимают из бани и быстро приливают столько холодной воды, чтобы до мерной черты оставался объем не более 10 - 15 см3. Содержимое колбы охлаждают в проточной воде до температуры 20 °С.
Белковые вещества в растворе осаждают добавлением 2 см3 раствора калия железистосинеродистого (150 г/дм3) и после перемешивания 2 см3 раствора цинка сульфата (150 г/дм3). Затем мерную колбу в течение 10 - 15 мин выдерживают при комнатной температуре, доводят дистиллированной водой до метки, перемешивают и в течение 5 мин дают отстояться. Взамен обоих указанных реактивов в случае их отсутствия для осаждения белков и осветления раствора в колбу приливают 5 см3 раствора аммония молибдата (100 г/дм3), или 5 см3 раствора фосфорно-вольфрамовой кислоты (40 г/дм3), или 3 см3 раствора натрия молибдата (165 г/дм3). При использовании молибдатов в качестве осадителей белков рекомендуется избегать попадания солнечных лучей на реактивы. Содержимое колбы через сухой складчатый фильтр фильтруют в сухую коническую колбу, первые несколько капель фильтрата отбрасывают. Прозрачным фильтром заполняют трубку поляриметра и в поляриметре измеряют оптическое вращение. Угол вращения испытуемого раствора в трубке поляриметра измеряют 5 раз.
До начала и после каждого второго измерения производят контроль установки поляриметра на нуль. Средняя величина 5 измерений служит исходной величиной для дальнейших вычислений массовой доли крахмала.
Обработка результатов
Массовую долю крахмала (X) в процентах вычисляют по следующим формулам. При использовании сахариметра с нормальной шкалой:
Х = К´а
или при использовании поляриметра с круговой шкалой:
![]() ,
где
,
где
![]() - переводной коэффициент, который при длине трубки 2
дм равен 1,9;
- переводной коэффициент, который при длине трубки 2
дм равен 1,9;
а -
показатель сахариметра или поляриметра в градусах шкалы (переводные
коэффициенты ![]() для
длины трубки 1 дм умножают на 2).
для
длины трубки 1 дм умножают на 2).
Метрологические характеристики
Допустимые расхождения между результатами двух параллельных определений (r) не должны превышать значений:
r = 0,03 + 0,04×Х1, но не более 0,5 % абсолютного содержания крахмала в БАД, где
Х1 - среднее арифметическое двух параллельных определений.
Допустимые расхождение между результатами измерений, выполненных в двух разных лабораториях, не должно превышать значений:
R = 0,05 + 0,06×Х2, но не более 1,2 % абсолютного содержания крахмала в продукте, где
Х2 - среднее значение результатов измерений, выполненных в двух разных лабораториях.
2. Определение содержания и состава углеводов
2.1. Определение состава углеводов с помощью метода ГЖХ
Метод основан на переводе углеводов типа глюкозы, фруктозы, арабинозы, ксилозы, галактозы, сахарозы, мальтозы, лактозы, рафинозы, сорбита и инозита в БАД в триметилсилильные производные с последующим их газохроматографическим анализом.
Специфические реактивы, материалы и аппаратура
Гексан х.ч.
Гексаметилдисилазан.
Трифторуксусная кислота или триметилхлорсилан.
Пиридин х.ч., безводный.
Ксилит х.ч. или инозит.
Свинец уксуснокислый х.ч.
Неподвижные фазы для ГЖХ SE-30, OV-17, ХЕ-60, СКТФТ-50.
Инертный носитель для ГЖХ: хромосорб-W DMCS, хроматон-N DMCS.
Газовый хроматограф с пламенно-ионизационным детектором и устройством для программирования температуры.
Стеклянная насадочная колонка длиной 2 - 3 м и диаметром 3 - 4 мм или капиллярная колонка длиной 25 - 30 м с нанесенной фазой.
Микрошприц емкостью 1,0 - 10 мкл.
Роторный испаритель.
Подготовка образцов
БАД с низким содержанием лимонной кислоты (менее 1,5 %)
Содержание лимонной кислоты устанавливается по «Таблицам химического состава». Навеску гомогенизированной пробы БАД с высоким содержанием сухих веществ (более 60 %) наносят на полиэтиленовую пластинку в количестве 50 - 150 мг и взвешивают на аналитических весах с точностью до 0,0001 г. На этой же пластинке взвешивают 5 - 10 мг ксилита. Пластинку помещают в круглодонную колбу емкостью 10 - 25 см3 и силилируют без обезвоживания с использованием трифторуксусной кислоты. При использовании триметилхлорсилана содержимое колбы упаривают досуха на роторном испарителе при 60 °С под вакуумом. В случае медленного упаривания в колбу добавляют 1 см3 бензола. После упаривания проводят силилирование.
Жидкие БАД отмеряют пипеткой в количестве 0,5 - 1,0 см3 и помещают в круглодонную колбу емкостью 10 - 25 см3, снабженную шлифом. Туда же на полиэтиленовой пластинке вносят навеску 5 - 10 мг ксилита, взвешенного с точностью до 0,0001 г. Содержимое колбы упаривают досуха на роторном испарителе.
БАД с содержанием лимонной кислоты более 1,5 %
Для исключения наложения пика ТМС-производного лимонной кислоты на ТМС-производное b-глюкозы, лимонную кислоту осаждают уксуснокислым свинцом. Навеску средней гомогенизированной пробы БАД (содержащие лимонную кислоту, как пищевую или консервирующую добавку) в количестве 1 - 15 г взвешивают с точностью до 0,001 г в конической колбе емкостью 100 см3, приливают 30 см3 75 - 80 % раствора этанола и экстрагируют 30 мин при температуре 60 - 70 °С. Используют водяную баню и обратный холодильник. Экстракцию повторяют трижды, экстракты объединяют.
Колбу доводят до метки дистиллированной водой. В центрифужную пробирку на 10 см3 вносят пипеткой 5 см3 отфильтрованного экстракта, 0,5 см3 насыщенного раствора уксуснокислого свинца. Осадок центрифугируется. 1 см3 надосадочной жидкости переносят в шлифную круглодонную колбу емкостью 10 - 25 см3. Туда же на полиэтиленовой пластинке вносят навеска ксилита 5 - 10 мг. Содержимое колбы упаривается на роторном испарителе при 60 °С под вакуумом.
Получение триметилсилильных (ТМС) производных
Способ с использованием трифторуксусной кислоты
В подготовленную навеску образца приливают точно 1,0 см3 пиридина, 0,9 см3 гексаметилдисилазана, 0,1 см3 трифторуксусной кислоты, плотно закрывают и энергично встряхивают в течение 30 с. Вначале наблюдают расслоение жидкости на 2 фазы, при этом нижний слой незначителен. По мере стояния раствора в течение 20 - 30 мин это расслоение исчезает и начинает выделяться аммиак, что указывает на успешное протекание реакции силилирования. После прекращения выделения аммиака раствор выдерживают 12 ч при комнатной температуре или 1 ч при 60 °С. Длительно сохраняющееся расслоение, исчезающее только при нагревании, говорит о том, что реакция силилирования прошла не полностью из-за высокого содержания влаги (более 40 %) или повышенного содержания углеводов. В этом случае подготовку пробы повторяют, уменьшив при этом навеску или увеличив время высушивания на роторном испарителе.
Способ с использованием триметилхлорсилана
В подготовленную навеску образца приливают точно 1,0 см3 пиридина, 0,2 см3 гексаметилдисилазана и 0,1 см3 триметилхлорсилана, встряхивают в течение 1 мин и нагревают в термостате 45 мин при 60 °С, затем хроматографируют.
Для упрощения идентификации и количественных расчетов (если не требуется знания аномерного состава сахаров) определение сахаров производят в виде ТМС-производных сахаров. В подготовленную пробу образца добавляют 100 г гидроксиламина солянокислого, приливают 10 см3 пиридина и выдерживают в термостате при 80 °С в течение 2 ч. Охлаждают и далее приливают силилирующие реагенты.
Газохроматографический анализ
Подготовка хроматографической колонки
Условия: Стеклянная колонка, наполненная сорбентом с 3 - 5 % неподвижной жидкой фазы длиной 2 м и внутренним диаметром 3 мм. Температуру колонки программируют от 125 до 270 °С со скоростью 4 °С в 1 мин, температуру испарителя 250 °С, расход газа-носителя 40 см3/мин, температура пламенно-ионизационного детектора 250 °С.
Газохроматографическое определение
1,0 мкл пиридинового раствора триметилсилильных производных углеводов вводят в испаритель хроматографа и элюируют из колонки газом-носителем. Идентификацию индивидуальных триметилсилильных производных проводят по времени удерживания триметилсилильных производных сахаров-метчиков и методом добавки.
Приготовление калибровочных растворов
Сахара, имеющие a- и b-аномеры
Навеску ксилита и навеску определяемого сахара, взятую с точностью до 0,0001 г, помещают в коническую колбу и заливают дистиллированной водой до полного растворения углеводов. Раствор выдерживают в течение суток. Аликвотную часть раствора отбирают пипеткой, помещают в круглодонную колбу и упаривают досуха, после чего проводят силилирование.
Сахара, не имеющие аномеров
Сахара, не имеющие аномеров, силилируют без предварительного растворения.
Обработка результатов
Массовую долю отдельных сахаров (в %) в навеске БАД определяют по формуле:
![]() ,
где
,
где
С1 - содержание отдельного сахара в навеске, %;
k1 - поправочный коэффициент данного сахара;
С - масса навески стандарта, мг;
Q - навеска образца;
Ас - площадь пика стандарта в относительных единицах;
a1 - площадь пика данного сахара в относительных единицах.
Расчет площадей хроматографических пиков сахарозы и a-лактозы при их совместном присутствии, в пробе
В виду того, что a-лактоза и сахароза выходят на хроматограмме одним пиком, площадь пика a-лактозы определяют, исходя из соотношения площадей a- и b-лактозы в модельном соединении лактозы. За основу берется площадь пика b-лактозы. Площадь пика сахарозы определяют вычитанием от суммарного пика сахарозы пика a-лактозы, рассчитанного из пика b-лактозы. За окончательный результат принимают среднеарифметическое (X) результатов двух параллельных определений.
Окончательный результат округляют до трехзначной цифры.
Метрологические характеристики
Относительное допустимое расхождение между результатами двух параллельных определений, выполненных в одной лаборатории, по отношению к среднему арифметическому значению (Rr), и относительное допустимое расхождение между результатами испытаний, выполненных в двух разных лабораториях, по отношению к среднему арифметическому значению (RR) зависят от содержания сахаров в продукте.
При содержании отдельных сахаров типа глюкозы, фруктозы, арабинозы, ксилозы, галактозы, сахарозы, лактозы, мальтозы, рафинозы, инозита и сорбита в продукте в пределах от 1 до 5 %:
· значения Rr = 20 %, RR = 40 % для каждого сахара; при содержании отдельного сахара в продукте свыше 5 %;
· значения Rr = 10 %, RR = 25 % для каждого сахара.
2.2. Определение состава углеводов с помощью метода ВЭЖХ
Настоящий метод основан на выделении, разделении и количественном определении с помощью высокоэффективной жидкостной хроматографии моно-, ди-, олигосахаров, а также сахарных спиртов. Он распространяется на определение глюкозы, фруктозы, арабинозы, фукозы, галактозы, ксилозы, лактозы, лактулозы, мальтозы, мальтотриозы, маннозы, сахарозы, раффинозы, стахиозы, сорбита, инозита, мальтита, ксилита, изомальта, лактитола добавленных в БАД.
Приборы и реактивы
Прибор для проведения хроматографии методом ВЭЖХ, состоящий из насоса, детектора-рефрактометра, колонки 250 мм ´ 4 мм, Сепарон SGX NH2 или аналогичная.
Компьютер с программным обеспечением по приему и обработке хроматографических данных.
Ротационный испаритель, водяная баня, обратный холодильник, две стеклянные колонки 7 - 10 см (внутренний диаметр 10 мм).
Ацетонитрил СН3СN для хроматографии.
Этиловый спирт 80 %.
Ионообменная смола Дауэкс 50´4, 200 - 400 меш (Н+-форма).
Ионообменная смола Дауэкс 1´8, 200 - 400 меш (формиатная форма).
2Н NaOH, 3Н HCl, 3Н формиат натрия, бидистиллированная вода.
0,1 %-ный раствор азотнокислого серебра.
Изопропиловый спирт.
Экстракция
Углеводы извлекают из БАД 80 % этиловым спиртом с учетом естественной влаги. Навеску БАД заливают в колбе в соотношении 1:4 по сухим веществам 96 % этанолом и необходимым количеством воды с расчетом, чтобы общая концентрация спирта была в пределах 80 - 82 %. Колбу нагревают с обратным холодильником на водяной бане при 70 - 80 °С в течение 15 мин. Затем спиртовую вытяжку отфильтровывают. К остатку приливают опять раствор 80 %-ного спирта и экстракцию повторяют в тех же условиях. Всего углеводы экстрагируют из продукта 3 - 4 раза.
Спиртовые экстракты объединяют, спирт отгоняют на ротационном испарителе при температуре не выше 40 °С.
Очистка экстракта
Предназначенные для определения экстракты нейтральных сахаров содержат также вещества, имеющие заряд (аминокислоты, пептиды и др.) Чтобы освободить нейтральные сахара от этих примесей, экстракты пропускают через колонки с дауэксом 50´4, непосредственно соединенную с колонкой, содержащей дауэкс 1´8.
Подготовка дауэкс 50´4, 200 - 400 меш.
Смолу последовательно промывают на бюхнеровской воронке большими объемами 2н NaOH, дистиллированной водой, 3н HCl и опять водой. Избыток воды удаляют путем длительного отсасывания насосом. Готовят суспензию смолы в дистиллированной воде (1:1) и используют ее для заполнения колонки.
Ионнообменная способность для дауэкса 50´4 в сухом виде составляет 5,1 мэкв/см3 и во влажном 1,7 мэкв/см3 соответственно.
Подготовка дауэкс 1´8, 200 - 400 меш.
Для работы нужна формиатная форма смолы. Последнюю готовят из дауэкса 1´8 (СГ-форма), пропуская через смолу на бюхнеровской воронке 3н формиат натрия до тех пор, пока проба на СГ не станет отрицательной (проба с азотнокислым серебром). После этого смолу промывают большими объемами дистиллированной воды и заполняют колонку. Ионнообменная способность для дауэкса 1´8 в сухом виде составляет 3,2 мэкв/см3 и во влажном виде 1,4 мэкв/см3.
С двух колонок собирают элюент и, если необходимо, его концентрируют в роторном испарителе. Чтобы упарить воду и не поднимать температуру, к образцу добавляют этанол. Упаренный образец переносят в мерную посуду и замеряют объем и хроматографируют.
Анализ ВЭЖХ
Подготовка колонки
Для этого колонку промывают 40 см3 изопропанола, затем 80 см3 деионизированной воды, после чего уравновешивают колонку подвижной фазой до стабильной нулевой линии.
Условия ВЭЖХ
Подвижная фаза: ацетонитрил-вода (77:23). Смесь дегазируют на роторном испарителе. Подвижная фаза для мальтотриозы, стахиозы и рафинозы: ацетонитрил-вода (60:40).
Детектирование - рефрактометр. Скорость потока 2,5 см3/мин. Стандартный раствор углевода 10 мг/см3 предварительно высушивают в сушильном шкафу при 105 °С до постоянного веса в стеклянных бюксах. Обработка хроматографических данных по программе МультиХром для Windows или аналогичной. Стандартный образец и испытываемую пробу хроматографируют не менее трех раз. Ошибка метода ±5 % колонку.
Определение содержания глюкозы в смеси с сорбитом
При описанных выше условиях хроматографирования время удерживания глюкозы и сорбита одинаковое и на хроматограмме они выходят единым пиком. В таких случаях количество глюкозы определяют глюкозооксидазным методом с использованием соответствующего ферментативного набора. Проведение анализа описано в соответствующем наборе. Количество сорбита определяют расчетным способом, как разницу суммарного содержания глюкозы и сорбита (по хроматограмме) и глюкозы (полученной ферментативным методом). В связи с разницей рефрактометрического индекса глюкозы и сорбита в расчетную формулу вводят поправочный коэффициент:
![]() ,
где
,
где
К - коэффициент пропорциональности, взятый из программы МультиХром.
3. Определение содержания пектина
Образец измельчают, взвешивают около от 1 до 5 г (в зависимости от того, сколько содержится пектина в образце), переносят в стеклянную колбу, заливают кипящей водой, в которую добавлена концентрированная соляная кислота из расчета 0,8 см3 на 250 см3 дистиллированной воды. Смесь нагревают при перемешивании в течение 30 мин при 95 - 100 °С. Охлаждают до температуры ниже 25 °С, чтобы свести к минимуму тепловое разрушение пектина, и фильтруют на воронке Бюхнера через капроновую ткань с отсасыванием. Если смесь плохо фильтруется, ее центрифугируют при 5000 об/мин в течение 30 мин. Экстракцию проводят дважды, экстракты и промывную воду объединяют, измеряют количество и к охлажденному раствору пектина прибавляют 1,5 объема этанола (можно использовать изопропиловый спирт или ацетон), содержащего 2 см3 концентрированной соляной кислоты (уд. вес 1,19) на 1 л. Смесь перемешивают вручную медленно и тщательно и оставляют стоять на 30 мин с тем, чтобы пектин всплыл на поверхность. Большую часть не содержащего пектина раствора можно отделить сифонированием. Осадок пектина отделяют центрифугированием, промывают спиртом до тех пор, пока реакция с нитратом серебра на ион-хлорид в промывных растворах будет отрицательной. Осадок сушат в термостате при 60 °С до постоянного веса.
Содержание пектина определяют гравиметрически.
Ошибка метода 5 %. Метод не пригоден для БАД, содержащих большие количества (более 30 %) сахарозы.
4. Методы определения содержания редуцирующих веществ, общего сахара и сахарозы
4.1. Колориметрический метод
Метод основан на колориметрировании избытка щелочного раствора гексацианоферрата (III) калия после реакции с редуцирующими сахарами объекта исследования. При этом гексацианоферрат (III) восстанавливается до гексацианоферрата (II), что ведет к ослаблению окраски, так как К3[Fе(СN)6] окрашен значительно интенсивнее, чем К4[Fe(CN)6].
Специфические реактивы
Глюкоза кристаллическая гидратная, х.ч., по ГОСТу 975-88.
Гексацианоферрат (III) калия, х.ч. по ГОСТу 4206-75.
Гидроксид калия, ч.д.а. по ГОСТу 24363-80 или гидроксид натрия, ч.д.а. по ГОСТу 4328-77, раствор с концентрацией NaOH (или КОН) 2,0М и 1,25М.
Кислота хлороводородная, х.ч. по ГОСТу 3118-77, раствор с концентрацией 1М.
Натрия хлорид, х.ч., по ГОСТу 4233-77.
Метиленовый голубой, индикатор, 1 г растворяют в 100 см3 дистиллированной воды и фильтруют.
Сахароза, х.ч. по ГОСТу 5833-75 или сахар-рафинад по ГОСТу 22-78.
Цинка сульфат, х.ч., по ГОСТу 4174-77, раствор с концентрацией 0,5М.
Внимание! Перед проведением анализа необходимо в обязательном порядке проверить вместимость мерной посуды и пипеток по I классу точности!
Подготовка к анализу
Приготовление раствора гексацианоферрата (III) калия - основной реактив. Взвешивают 8 г К3[Fе(СN)6] и 20 г NaOH (или 28 г КОН). Отдельно растворяют в небольшом количестве дистиллированной воды. Затем оба раствора сливают в мерную колбу вместимостью 1000 см3 и доводят до метки дистиллированной водой. Раствор готов к использованию через сутки. Раствор можно хранить в склянке из темного стекла в течение 2 мес.
Приготовление стандартного раствора глюкозы
1,6000 г безводной глюкозы взвешивают с точностью до 0,0002 г и растворяют в мерной колбе вместимостью 1000 см3. Предварительно глюкозу выдерживают в эксикаторе над свежепрокаленным хлоридом кальция в течение 3 сут. После растворения навески раствор в колбе доводят до метки. Если раствор готовят на месяц, необходимо внести в колбу 150 г хлорида натрия и хранить в холодильнике.
Построение градуированного графика
В 6 конических колб вместимостью 100 см3 вносят пипеткой по 25 см3 щелочного раствора гексацианоферрата (III) калия и по 7,0; 7,5; 8,0; 8,5; 9,0; 9,5 см3 стандартного раствора глюкозы. Из бюретки соответственно приливают 9,0; 8,5; 8,0; 7,5; 7,0 см3 дистиллированной воды, тем самым доводят объем жидкости в каждой колбе до 41 см3. Содержимое каждой колбы нагревают до кипения за 3 мин и кипятят в течение 1 мин, закрыв часовым стеклом. Затем охлаждают и измеряют оптическую плотность на ФЭК со светофильтром, имеющим lэф = 440 нм (синий светофильтр). Раствором сравнения служит дистиллированная вода. Кювету подбирают такого размера, чтобы оптическая плотность была в пределах 0,3 - 0,6 для раствора, содержащего 8,5 см3 раствора глюкозы (10, 20 или 30 мм). Оптическую плотность определяют в каждом растворе не менее 3-х раз и из полученных данных берут среднее арифметическое значение. Строят график зависимости величины оптической плотности от концентрации глюкозы в растворе. Полученный график используют для определения содержания общего сахара, восстанавливающих сахаров и сахарозы.
Приготовление исследуемого раствора
Объект исследования тщательно измельчают в ступке. Затем готовят водную вытяжку объекта исследования. Массу навески М в граммах рассчитывают по формуле:
![]() ,
где
,
где
V - вместимость колбы, см3;
Р - предполагаемое содержание общего сахара в объекте исследования, %;
С - оптимальная для данного метода концентрация сахаров в водной вытяжке на 100 см3 (принимают равной 0,16 г).
Растворение навески и осаждение несахаров проводят следующим образом. Образец измельченного исследуемого БАД взвешивают с погрешностью не более 0,01 г из такого расчета, чтобы в 100 см3 полученного раствора содержалось 0,3 - 0,4 г редуцирующих веществ. Массу навески М в граммах вычисляют по формуле:
![]() ,
где
,
где
С - оптимальное содержание редуцирующих веществ в 100 см3 раствора навески, г;
V - вместимость мерной колбы, см3;
Р - предполагаемая массовая доля редуцирующих веществ в исследуемом изделии, %.
Навеску растворяют в стакане в дистиллированной воде, нагретой до 60 - 70 °С. Если БАД растворяется без остатка, то полученный в стакане раствор охлаждают и переносят в мерную колбу вместимостью 250 см3, доводят объем раствора до метки дистиллированной водой и хорошо перемешивают. Если БАД в своем составе имеет вещества, нерастворимые в воде (мешающие несахара - белки, жиры, пектины, крахмал и т.д.), то навеску в стакане переносят в мерную колбу вместимостью 250 см3, смывая нерастворимые частицы в колбу дистиллированной водой примерно до половины объема колбы. Органические кислоты, содержащиеся в навеске, нейтрализуют раствором углекислого натрия до рН 7,0, применяя для контроля универсальную индикаторную или лакмусовую бумагу. Колбу помещают в водяную баню, нагретую до 60 °С, при этой температуре, временами взбалтывая, выдерживают в течение 15 мин. Охладив раствор до комнатной температуры, осаждают мешающие несахара, прибавляя к раствору в колбе 10 см3 1M раствора сульфата цинка, если масса навески была менее 5 г, и 15 см3, если масса навески была более 5 г, и такой объем 1М раствора гидроксида натрия, который установлен отдельным опытом при титровании соответствующего объема раствора сульфата цинка с фенолфталеином.
Содержимое колбы взбалтывают, доводят дистиллированной водой до метки, перемешивают и фильтруют в сухую колбу, которую предварительно ополаскивают 1 - 2 раза небольшой порцией фильтрата.
Допускается осаждение несахаров другими осадителями: растворами ацетата свинца и фосфата (или сульфата) натрия и растворами гексацианоферрата (II) калия и ацетата цинка. При осаждении несахаров ацетатом свинца к охлажденному до комнатной температуры раствору прибавляют мерным цилиндром 7 см3 раствора ацетата свинца, хорошо перемешивают и оставляют стоять 5 мин. Появление прозрачного слоя жидкости над осадком указывает на полноту осаждения, в противном случае вносят дополнительно по каплям раствор ацетата свинца до появления прозрачного слоя жидкости. Затем в эту же колбу для удаления избытка ацетата свинца вносят 18 - 20 см3 раствора фосфата (или сульфата) натрия.
Содержимое колбы взбалтывают, осадку дают отстояться. Для осаждения избытка ацетата свинца фосфатом натрия достаточно 10 мин. При осаждении сульфатом натрия при мутном растворе жидкость отстаивается 24 ч. После отстаивания проверяют полноту осаждения, приливая по стенке горлышка колбы нескольких капель раствора фосфата или сульфата натрия. При помутнении жидкости прибавляют дополнительно один из указанных выше растворов (1 - 2 см3), затем содержимое колбы взбалтывают, дают отстояться и снова проверяют полноту осаждения. При отсутствии помутнения в месте соприкосновения жидкостей содержимое колбы доводят до метки дистиллированной водой, перемешивают и фильтруют в сухую колбу.
При осаждении несахаров раствором гексацианоферрата (II) калия к охлажденному до комнатной температуры раствору прибавляют пипеткой 2 см3 раствора гексацианоферрата (II) калия, взбалтывают, добавляют 2 см раствора ацетата цинка, снова взбалтывают и дают отстояться 5 мин. Если раствор над осадком остается мутным, добавляют большее количество указанных растворов в равных объемах. Содержимое колбы доводят водой до метки, перемешивают и фильтруют в сухую колбу. Во всех случаях фильтрат должен быть прозрачным.
При анализе окрашенных БАД (сокосодержацих и т.п.) необходимо связать красящие вещества и нейтрализовать органические кислоты раствором 1М гидроксида натрия или 1М гидроксида калия и связать дубильные вещества ацетатом свинца. Для этого добавляют к пробе по капле раствор карбоната натрия или 1М раствор гидроксида натрия до слабокислой или нейтральной реакции и 30 %-ный раствор ацетата свинца. После тщательного перемешивания и отстаивания добавляют по каплям раствор сульфата натрия до прекращения образования осадка, для связывания избытка ацетата свинца. Доводят водой до метки, отстаивают и фильтруют.
Проведение анализа
Определение восстанавливающих сахаров
В коническую колбу вместимостью 100 - 150 см3 вносят 10 см3 раствора пробы, 6 см3 дистиллированной воды и затем 25 см3 щелочного раствора гексацианоферрата калия. Колбу нагревают до кипения за 3 мин, кипятят 1 мин, охлаждают и измеряют оптическую плотность при l = 440 нм. Раствор сравнения - дистиллированная вода. Кювету берут размером, аналогичным взятому для построения градуировочного графика. Если оптическая плотность раствора не попадает в интервал 0,3 - 0,6, необходимо взять меньшую аликвоту пробы или поменять разведение, сохраняя постоянный объем жидкости в реакционной колбе, равным 41 см3. Результаты вычисляют, пользуясь градуировочным графиком и ниже приведенной формулой.
Определение общего сахара
Для определения общего сахара проводят гидролиз сахарозы. В реакционную коническую колбу вместимостью 100 - 150 см3 отмеривают пипеткой 10 см3 приготовленной вытяжки объекта и 4 см3 1М раствора хлороводородной кислоты. Колбу ставят на электроплитку, жидкость доводят до кипения и кипятят 1 мин, охлаждают до комнатной температуры. Затем в колбу вносят 2 см3 2М раствора гидроксида натрия (или калия) для нейтрализации кислоты и затем 25 см3 щелочного раствора гексацианоферрата (III) калия. Содержимое колбы доводят до кипения и кипятят 1 мин. После охлаждения заполняют полученным раствором кювету и определяют оптическую плотность так же, как и при снятии градуировочного графика. Так как при значении оптической плотности в интервале 0,3 - 0,6 получаются более точные результаты, то при других значениях оптической плотности анализ повторяют, соответственно изменив объем вводимой водной вытяжки объекта исследования, добавив в воду так, чтобы общий объем оставался равным 10 см3.
Обработка результатов
Расчет содержания сахара
Содержание сахара Хрс (в %), выраженное в глюкозе, вычисляют по формуле:
![]() ,
где
,
где
М - количество глюкозы, найденное по градуировочному графику, мг;
V - объем исследуемого раствора, приготовленного из навески, м3;
V1 - объем раствора, взятый для реакции с гексацианоферратом калия, см3;
т - масса навески объекта исследования, мг.
Для определения содержания общего сахара, выраженного в сахарозе, результат, полученный по формуле, умножают на 0,95. Если калибровку проводят с использованием раствора гидролизованной сахарозы, результат сразу выражают в % сахарозы. Для расчета содержания сахарозы из данных анализа общего сахара, выраженного в глюкозе, вычитают результат анализа содержания восстанавливающих сахаров и разницу умножают на коэффициент 0,95.
Вычисления проводят до второго десятичного знака. За результат измерения принимают среднее арифметическое значение результатов двух параллельных определений (X1) и выражают целым числом с одним десятичным знаком.
Метрологические характеристики
Допустимые расхождения между результатами двух параллельных (r) определений не должны превышать:
r = 0,02 + 0,03×Х1, но не более 0,5 % абсолютного содержания сахара в БАД, где
X1 - среднее арифметическое значение двух параллельных определений.
Допустимое расхождение между результатами измерений, выполненных в двух разных лабораториях, не должно превышать:
R = 0,04 + 0,05×Х2, но не более 1,0 % абсолютного значения содержания сахара в продукте, где
Х2 - среднее значение результатов измерений, проведенных в двух разных лабораториях.
4.2. Титрометрический метод
Метод основан на титровании избытка гексацианоферрата калия стандартным раствором глюкозы в присутствии метиленового голубого до полного обесцвечивания.
Подготовку к анализу проводят также как и в колориметрическом методе.
Проведение анализа
Определение восстанавливающих сахаров
В коническую колбу вместимостью 100 - 150 см3 вносят 10 см3 раствора пробы, 25 см3 щелочного раствора гексацианоферрата калия. Колбу нагревают до кипения за 3 мин, кипятят ровно 3 мин и вводят 2 капли метиленового голубого, дотитровывают без прекращения кипячения раствором глюкозы до исчезновения синей окраски. Содержание сахаров определяют по формуле, представленной выше.
Холостой опыт проводят в тех же условиях, отбирая вместо 10 см3 раствора пробы 10 см3 стандартного раствора глюкозы.
Количество стандартного раствора глюкозы V1, пошедшее на титрование 25 см3 щелочного раствора гексацианоферрата (III) калия, суммируется как 10 см3 глюкозы, взятые на анализ, и объем глюкозы, который пошел на дотитровывание.
Определение общего сахара
Проводят предварительный гидролиз сахарозы. Затем вносят 25 см3 щелочного раствора гексацианоферрата (III) калия и далее проводят кипячение и титрование, как и для определения восстанавливающих сахаров.
Расчет общего сахара
Содержание общего сахара (Хос) в %, выраженное в глюкозе, вычисляют по формуле:
![]() ,
где
,
где
V1 - количество стандартного раствора глюкозы, пошедшее на титрование 25 см3 щелочного раствора гексацианоферрата (III) калия в холостом опыте, см3;
V2 - количество стандартного раствора глюкозы, пошедшее на дотитрование, см3;
1,6 - количество глюкозы в 1 см3 раствора, мг;
Vк - вместимость мерной колбы, используемой для приготовлении водной вытяжки, см3;
М - масса навески объекта исследования, мг.
Для получения содержания общего сахара, выраженного в сахарозе, результат, полученный по формуле, умножают на 0,95. Если калибровку проводят с использованием раствора гидролизованной сахарозы, результат сразу выражают в % сахарозы. Для расчета содержания сахарозы из данных анализа общего сахара, выраженного в глюкозе, вычитают результат анализа содержания восстанавливающих сахаров и разницу умножают на коэффициент 0,95.
Вычисления проводят до второго десятичного знака. За результат измерения принимают среднее арифметическое значение результатов двух параллельных определений (X1) и выражают целым числом с одним десятичным знаком.
Метрологические характеристики фотометрического и титрометрического методов
Допустимые расхождения между результатами двух параллельных (r) определений не должны превышать:
r = 0,03 + 0,035×X1, но не более 0,5 % абсолютного содержания сахара в БАД, где
X1 - среднее арифметическое значение двух параллельных определений.
Допустимые расхождение между результатами измерений, выполненных в двух разных лабораториях, не должно превышать:
r = 0,04 + 0,05×X2, но не более 1,0 % абсолютного содержания сахара в БАД, где
X2 - среднее значение результатов измерений, проведенных в двух разных лабораториях.
5. Определение содержания нерастворимых и растворимых пищевых волокон (ферментативный метод)
Сущность метода заключается в гидролизе и удалении белковых и крахмалистых веществ ферментами, аналогичными ферментам пищеварительного тракта человека из БАД на растительной основе. Метод позволяет определять растворимые (в этиловом спирте) и нерастворимые пищевые волокна, отличающиеся физиологическим действием.
Специфическая аппаратура, материалы, реактивы
Весы лабораторные общего назначения с наибольшим пределом взвешивания 200 г второго класса точности по ГОСТ 24104-80.
Шкаф сушильный лабораторный.
Спектрофотометр СФ-26 или другой с аналогичными характеристиками.
Водяная баня любого типа, обеспечивающая температуру нагрева 37±0,2 °С, 40±0,2 °С, 60±0,2 °С, 100 °С, или термостат.
Прибор для определения рН среды с диапазоном измерений 0 - 14 с погрешностью измерения ±0,1 ед. рН.
Воронки стеклянные с пористым фильтром № 40, № 100 по ГОСТ 25336-82.
Спирт этиловый по ГОСТ 5962-67 или ГОСТ 18300-72 и раствор с массовой долей этилового спирта 700 г/дм3.
Панкреатин, фармзавод Лейрас, А/О Хухтамяки, медицинский препарат, активность которого гарантирована в пределах установленных сроков хранения.
Глюкоамилаза очищенная по ТУ 64-13-18-88.
Гемоглобин бычий окисленный лиофилизированный МБ по ТУ 6-09-10-656-77.
Протеаза № Р-3910, Sigma Chemical Co. (хранится только в холодильнике) или другой протеолитический ферментный препарат аналогичной активности, например, пепсин А из слизистой оболочки желудка свиньи, активность которого устанавливают следующим образом. Сначала готовят 2 %-ный раствор гемоглобина. Для этого 300 мг гемоглобина растворяют в 12,9 см3 дистиллированной воды (до полного растворения), приливают 2,1 см3 раствора соляной кислоты концентрации 0,3М.
Определение активности проводят следующим образом: 68,5 мг пепсина растворяют в 10 см3 раствора соляной кислоты концентрации 0,03М; 0,2 см3 полученного раствора пепсина приливают к 1 см3 субстрата гемоглобина, предварительно нагретого на водяной бане в течение 5 мин при температуре 37 °С. Инкубируют смесь при той же температуре в течение 10 мин (по секундомеру). Реакцию останавливают путем приливания 5 см3 раствора соляной кислоты концентрации 100 г/дм3. Смесь выдерживают на водяной бане еще 5 мин. Фильтруют через плотный бумажный фильтр (фильтраты должны быть абсолютно прозрачными). Одновременно с опытными готовят контрольную пробу. Для этого к 1 см3 субстрата гемоглобина приливают 0,2 см3 раствора соляной кислоты концентрации 0,03М и 5 см3 10 % раствора трихлоруксусной кислоты, затем инкубируют смесь и фильтруют по предложенной выше схеме. Содержание пепсина определяют спектрофотометрически: оптическую плотность опытных и контрольных фильтратов измеряют против дистиллированной воды при длине волны 280 нм в кюветах 1 см на СФ-26 или другом с подобными характеристиками.
Построение калибровочного графика: готовят ряд растворителей рабочего стандарта пепсина с концентрацией фермента 50, 100, 150, 200, 250 мкг/см3. Для этого пользуются следующими данными, приведенными в табл. 9.
Таблица 9
Количество ферментов
|
50 |
100 |
150 |
200 |
250 |
|
|
Количество раствора рабочего стандарта пепсина, см3 |
0,1 |
0,2 |
0,3 |
0,4 |
0,5 |
|
Количество раствора соляной кислоты с концентрацией 0,03 М, см3 |
9,9 |
9,8 |
9,7 |
9,6 |
9,5 |
Во всех растворах, приготовленных по таблице, определяют содержание пепсина, как описано выше, и строят график, откладывая на оси ординат величины оптической плотности, а на оси абсцисс - концентрацию пепсина в мкг. По калибровочному графику определяют количество активного пепсина в исследуемом препарате. Результаты измерений выражают в протеиназных единицах (по гемоглобину), ПЕНВ. За протеиназную единицу принимают действие такого количества фермента, которое в данных условиях опыта и измерений за 1 мин приводит к увеличению оптической плотности фильтрата на единицу. Например, по предложенному калибровочному графику 250 мкг пепсина увеличивают оптическую плотность фильтрата на 0,5 за 10 мин, следовательно, 5 000 мкг пепсина приводит к увеличению оптической плотности на единицу за 1 мин (1 ПЕНВ = 5000 мкг или 1 мкг = 5´10-3 ПЕНВ).
Подготовка к испытанию
Приготовление фосфатного буферного раствора панкреатина
Готовят 0,05М фосфатный буфер рН 6,0 (0,875 г дигидрофосфата и 6,05 г натрия гидрофосфата растворяют в примерно 700 см3 дистиллированной воды в колбе на 1 дм3, затем доводят до метки дистиллированной водой).
Готовят раствор панкреатина с концентрацией панкреатина в 0,05М фосфатном буфере 5 мг/см3.
Приготовление 0,5 %-ного водного раствора глюкоамилазы
Растворяют 500 мг глюкоамилазы в 100 см3 дистиллированной воды.
Подготовка образцов к испытанию
Исследуемый сухой материал измельчают на лабораторной мельнице, просеивают через сито, собирая фракцию с размером частиц не более 1 мм; влажный материал гомогенизируют в гомогенизаторе или микроизмельчителе тканей.
При содержании жира в образцах более 5 % проводят обезжиривание. Для этого навеску образца, предназначенную для определения пищевых волокон, заливают трехкратным объемом петролейного эфира (к массе образца), перемешивают в течение 15 мин периодически и затем отстаивают не менее 1 мин. Прозрачный раствор петролейного эфира декантируют и повторяют экстракцию с тем же количеством эфира 2 раза. Обезжиренный образец высушивают на воздухе.
Ориентировочное содержание крахмала в образце устанавливают по справочным таблицам или другим любым приемлемым методом.
Проведение испытания
0,5 г тонкоизмельченного образца, взвешенного с точностью до 0,0001 г, помещают в химический стакан и суспендируют в 30 см3 дистиллированной воды при 40 °С в течение 1,5 ч.
Полученную суспензию после настаивания нагревают на кипящей водяной бане для клейстеризации крахмала 30 мин при условии, что температура суспензии достигает 90 °С.
После охлаждения смеси до комнатной температуры устанавливают рН 1,5 ± 0,1 раствором соляной кислоты концентрацией 5М, добавляют 100 мг пепсина (активность 1 мкг эквивалентна 5´10-3 ПЕНВ) и ставят на инкубацию при температуре 40 °С в течение 1 - 1,5 ч при периодическом помешивании. При недостаточной активности пепсина необходимо скорректировать его количество по вышеописанной методике при проведении ферментолиза.
Смесь охлаждают до комнатной температуры, затем устанавливают рН 6,8±0,1 раствором гидроксида натрия концентрацией 3М и приливают 10 см3 0,05 молярного буферного раствора панкреатина. Смесь инкубируют в течение 1 - 1,5 ч при помешивании при температуре 40 °С. Охлаждают полученную смесь до комнатной температуры, приливают 1,0 см3 водного раствора глюкоамилазы концентрацией 50 мг/см3 (рекомендуется использовать 100 единиц активности глюкоамилазы на расщепление 1 г крахмала), устанавливают рН 4,8±0,1 раствором соляной кислоты концентрацией 5М и инкубируют при температуре 40 °С в течение 12 ч.
Полноту гидролиза крахмала определяют йодной пробой. Для этого несколько капель смеси помещают на стекло, добавляют каплю йода в водном растворе йодида калия. О присутствии крахмала в препарате пищевых волокон судят по наличию окрашенных в синий цвет крахмальных зерен. В случае положительной реакции на крахмал проводят обработку смеси дополнительным количеством глюкоамилазы (приливают к смеси 5 см3 водного раствора глюкоамилазы концентрацией 5 мг/см3 и инкубируют 1 ч при аналогичных условиях: рН 4,8±0,1, температура 60 °С).
Полученную смесь фильтруют через предварительно доведенный до постоянной массы фильтр № 100 или предварительно высушенный и взвешенный обеззоленный фильтр. Нерастворимые пищевые волокна, оставшиеся на фильтре, промывают последовательно 25 см3 70 %-ного раствора этилового спирта (в 2 приема) и 25 см3 ацетона (в 2 приема). Фильтрат должен быть прозрачным.
Фильтр помещают в сушильный шкаф при температуре 105±1 °С и высушивают до постоянной массы. Фильтрат делят на 2 равные части. В одной части фильтрата осаждают растворимые пищевые волокна 4-кратным количеством 96 %-ного этилового спирта, взятого к объему фильтрата. Смесь оставляют для осаждения на 10 - 12 ч, затем фильтруют через доведенный до постоянной массы стеклянный пористый фильтр № 40 (при необходимости используют слабый вакуум) до получения прозрачного фильтрата. Остаток на фильтре промывают последовательно 25 см 70 %-ного раствора этилового спирта и 25 см3 ацетона, затем высушивают в сушильном шкафу при температуре 105±1 °С до постоянной массы.
В полученных препаратах нерастворимых пищевых волокон определяют содержание непереваренного белка по методу Кьельдаля и золы по ГОСТу 10847-74. Для корректировки данных по содержанию пищевых волокон от возможного осаждения остатка ферментов на растворимые и нерастворимые пищевые волокна параллельно проводят холостой опыт по вышеизложенной схеме, но без навески образца. Холостой опыт важно проводить при использовании новых партий ферментов.
Обработка результатов
Содержание пищевых волокон в БАД определяют по следующим формулам.
Для нерастворимых пищевых волокон:
![]() .
.
Для растворимых пищевых волокон:
![]() ,
где
,
где
X1 - содержание нерастворимых пищевых волокон, %;
Х2 - содержание растворимых пищевых волокон, %;
М - навеска образца, г;
m1 и М3 - масса остатка нерастворимых или растворимых пищевых волокон после высушивания соответственно, г;
m2 - масса остатка в холостом опыте после высушивания, г;
В - содержание белка в препарате пищевых волокон, г;
С - содержание золы в препарате пищевых волокон, г.
Общее содержание пищевых волокон (X) вычисляют суммированием величин X1 и Х2.
За окончательный результат определения массовой доли нерастворимых или растворимых пищевых волокон принимают среднее арифметическое результатов двух параллельных определений. Вычисление проводят до второго десятичного знака с последующим округлением до первого десятичного знака.
Метрологические характеристики
Относительное допустимое расхождение между результатами двух параллельных определений, выполненных в одной лаборатории, по отношению к среднему арифметическому значению (Rr) и относительное допустимое расхождение между результатами испытаний, выполненных в двух разных лабораториях, по отношению к среднему арифметическому значению (RR) приведены в табл. 10.
Таблица 10
Относительные допустимые внутрилабораторные (Rr) и межлабораторные (RR) расхождения результатов определения
|
Rr, % |
RR, % |
|
|
Растворимые |
15 |
20 |
|
Нерастворимые |
10 |
30 |
Глава 2
МЕТОДЫ ОПРЕДЕЛЕНИЯ МИКРОНУТРИЕНТОВ
I. Методы определения витаминов
1. Одновременное определение витаминов А, Е и каротиноидов в БАД методом высокоэффективной жидкостной хроматографии
Сущность метода
Метод определения витаминов А, Е и каротиноидов, включая b-каротин, a-каротин, ликопин, криптоксантин, лютеин, зеаксантин, при совместном присутствии в биологически активных добавках (витаминное драже, таблетки, порошки и кристаллические витаминные препараты, их растворы или суспензии в жирах) основан на экстракции микронутриентов органическим растворителем после щелочного омыления субстрата (1) или непосредственного растворения, упаривании полученного экстракта и переводе сухого остатка в другой растворитель, введении экстракта на ВЭЖХ колонку для хроматографического разделения и последующего определения с помощью флуоресцентного и спектрофотометрического детекторов (2). Определение массовой концентрации витаминов и каротиноидов основано на измерении площади (или высоты) пика при соответствующей каждому соединению длине волны детектирования после введения в хроматографическую систему анализируемых проб и градуировочных растворов.
Характеристика погрешности измерений
Настоящая методика измерений массовой концентрации жирорастворимых витаминов А, Е и каротиноидов в биологически активных добавках с вероятностью Р = 0,95 обеспечивает выполнение анализа с погрешностями ±15 % во всем диапазоне измерений.
Средства измерений, вспомогательные устройства, реактивы и материалы
Спектрофотометр, например, «СФ-46» с диапазоном измерения от 190 нм до 700 нм с допустимой абсолютной погрешностью измерений коэффициента пропускания не более 1 % по ТУ 3-3.1841-84; кюветы кварцевые рабочей длиной 10 мм.
Спектрофотометрический детектор для ВЭЖХ (типа «Джаско 870-UV», Япония) с проточной кюветой объемом 8 мкл (длина оптического пути 10 мм), уровнем относительной погрешности измерения не более 2 % с программируемой сменой длин волн, позволяющий проводить исследования в видимом и ультрафиолетовом диапазоне спектра (от 190 до 600 нм).
Спектрофлуориметрический детектор для ВЭЖХ (типа «Джаско» 821-FP) с проточной кварцевой кюветой объемом 16 мкл (длина оптического пути 10 мм), уровнем относительной погрешности измерения интенсивности флуоресценции не более 2 % с программируемой сменой длин волн, позволяющий проводить исследования в видимом и ультрафиолетовом диапазоне спектра возбуждения и эмиссии (от 220 - 650 нм).
Насос для ВЭЖХ (типа «Джаско» 880-PU), позволяющий осуществлять расход элюента 0,5 - 1,0 см3/мин с возможностью работы при давлении не менее 300 кг/см2, погрешностью поддержания скорости подачи растворителя 0,6 см3/мин не более 2,5 %.
Микрошприц (типа «Гамильтон», США) вместимостью 100 мм3 для ввода проб в жидкостный хроматограф.
Регистрирующее устройство: интегратор (типа «Шимадзу C-R6A», Япония) или самописец, позволяющий проводить измерение с погрешностью не более 0,5 %.
Дозатор автоматический типа ПЛ-01-200.
Колбы мерные 2-100-2, 2-50-2 по ГОСТ 1770-74Е.
Цилиндры вместимостью 100 см3 по ГОСТ 1770-74Е.
Пробирки П-4-10-14/23 ХС по ГОСТ 25336.
Линейка металлическая с ценой деления 1 мм по ГОСТ 427-75.
Вспомогательные устройства и материалы
Встряхиватель типа АВУ-6с по ТУ 64-1-2451-78.
Центрифуга ОПн-8-У4.2 по ТУ 5.375-4261-76.
Баллон с газообразным азотом квалификации «ос.ч» по ГОСТ 9293-74.
Реактивы
Метанол (СН3ОН) для жидкостной хроматографии «ос.ч» по ТУ 6-09-2192-85.
Ацетонитрил (СН3СN) для жидкостной хроматографии «ос.ч» по ТУ 6-09-14-2167-84.
Метилен хлористый (СН2Сl2), «ос.ч» по ТУ 6-09-14-2149-83.
Гексан (С6Н6), «хч» для хроматографии по ТУ 6-00-4521.
Спирт этиловый (C2H5OH) ректификованный технический по ГОСТ 18300-87.
Полностью транс-ретинол (С20Н30О) кристаллический, номер по каталогу 124769 фирмы «Мерк» (Германия).
Ретинола ацетат (С22Н32О2) по Госфармакопея, X изд., ст. 578.
Ретинола пальмитат (С26Н60О2) по ФС 42-2229-84.
DL-a-Токоферол (C29H50O2), номер по каталогу Т3251 фирмы «Сигма».
a-Токоферола ацетат (C31H52O2) по ФС 42-2495-87.
Каротиноиды: лютеин (С40Н56О2), ликопин (C40H56), a-каротин (C40H56), b-каротин (C40H56), соответствующие номера Х6250, L9879, С0251, С9750 по каталогу фирмы «Сигма» (США), зексантин С40Н56О2, b-криптоксантин (С40Н56О) фирмы «Рош» (Швейцария).
Калия гидроокись по ГОСТ 24363, ч.д.а.
Вода дистиллированная по ГОСТ 6709-72.
Средства измерения должны быть поверены в установленные сроки. Допускается использование других средств измерения, вспомогательного оборудования и реактивов, имеющих аналогичные или лучшие характеристики.
Условия выполнения измерений
Для предотвращения разрушения витаминов и каротиноидов анализ БАД и стандартов проводят в присутствии антиоксиданта - аскорбиновой кислоты, предохраняя пробы от попадания на них прямого солнечного света.
Подготовка к выполнению измерений
Приготовление растворов
1. Приготовление раствора гидроокиси калия с массовой долей 50 %.
50 г гидроокиси калия растворяют в 50 см3 дистиллированной воды.
2. Подвижная фаза для хроматографии
В мерную колбу на 500 см3 помещают 250 см3 ацетонитрила, 25 см3 дихлорметана и доводят объем смеси до метки метанолом. Раствор тщательно перемешивают. Срок хранения в темноте при 2 - 8 °С - 1 год.
Количественное определение витаминов А, Е и b-каротина
Количественное определение проводят методом абсолютной калибровки, для чего проводят определение градуировочного коэффициента.
Для определения градуировочного коэффициента готовят несколько (не менее 4) калибровочных растворов витамина А (полностью транс-ретинол кристаллический; ретинола ацетат по Госфармакопея, X изд., ст. 578; ретинола пальмитат по ФС 42-2229-84) с концентрацией от 0,3 до 3 мкг/см3, витамина Е с концентрацией от 2 до 20 мкг/см3 (DL-a-токоферол; a-токоферола ацетат по ФС 42-2495-87), b-каротина с концентрацией от 0,2 до 1,5 мкг/см3.
Приготовление градуированных растворов витамина А
Для приготовления градуировочных растворов из ретинола 0,01 г витамина А количественно переносят в мерную колбу вместимостью 100 см3, добавляют 50 см3 этанола. Содержимое колбы тщательно перемешивают до полного растворения кристаллов ретинола, после чего объем в колбе доводят до метки этиловым спиртом и содержимое колбы вновь тщательно перемешивают. После введения в колбу на 100 см3 аликвоты раствора витамина А в этаноле (0,3; 0,5; 1,0; 1,5; 2,0 см3) добавляют растворитель до метки.
Для приготовления калибровочных растворов из ацетата ретинола (пальмитата ретинола) 0,01 г витамина А помещают в коническую колбу вместимостью 100 см3, прибавляют 30 см3 этанола, 3 см3 50 % раствора гидроокиси калия и нагревают в течение 30 мин на водяной бане с обратным холодильником при температуре 63 - 68 °С. Содержимое колбы быстро охлаждают до комнатной температуры, количественно переносят 50 см3 воды в делительную воронку и извлекают 150 см3 гексана (3 раза по 50 см3) в течение двух минут. Объединенные гексановые экстракты промывают водой по 50 мл до исчезновения щелочной реакции промывных вод (по индикаторной бумаге). Промытые гексановые экстракты количественно переносят в колбу вместимостью 200 см3 и доводят объем раствора до метки тем же растворителем. Из полученного раствора отбирают 1, 2, 3, 4, 5 см3 раствора в мерные колбы вместимостью 100 см3, упаривают в токе азота, сухой остаток растворяют в этаноле и доводят объемы до метки тем же растворителем.
Приготовление градуировочных растворов витамина Е
Для приготовления градуировочных растворов из a-токоферола 0,02 г витамина Е количественно переносят в мерную колбу вместимостью 100 см3, добавляют 2 см3 дихлорметана. Содержимое колбы тщательно перемешивают до полного растворения токоферола, после чего объем в колбе доводят до метки этиловым спиртом и содержимое колбы вновь тщательно перемешивают. После введения в колбу на 100 см3 аликвоты раствора витамина Е в этаноле (1, 3, 5, 7, 10 см3) добавляют растворитель до метки.
Для приготовления градуировочных растворов из ацетата a-токоферола 0,02 г витамина Е количественно переносят в коническую колбу вместимостью 100 см3, добавляют 0,2 г аскорбиновой кислоты, 30 см3 этанола, 3 см3 50 %-ного раствора гидроокиси калия и нагревают в течение 30 мин на водяной бане с обратным холодильником при температуре 63 - 68 °С. Содержимое колбы быстро охлаждают до комнатной температуры, количественно переносят 50 см3 воды в делительную воронку и извлекают 150 см3 гексана (3 раза по 50 см3) в течение двух минут. Объединенные гексановые экстракты промывают водой по 50 мл до исчезновения щелочной реакции промывных вод (по индикаторной бумаге). Промытые гексановые экстракты количественно переносят в колбу вместимостью 200 см3 и доводят объем раствора до метки тем же растворителем. Из полученного раствора отбирают 2, 4, 6, 8, 10 см3 раствора в мерные колбы вместимостью 100 см3, упаривают в токе азота, сухой остаток растворяют в 1 см3 дихлорметана и доводят объемы до метки этанолом.
Приготовление градуировочных растворов каротиноидов
Для приготовления градуировочных растворов b-каротина 5 мг кристаллического b-каротина количественно переносят в мерную колбу вместимостью 50 см3, добавляют 1 мл дихлорметана. Содержимое колбы тщательно перемешивают до полного растворения кристаллов, после чего объем в колбе доводят до метки этанолом и содержимое колбы вновь тщательно перемешивают. После введения в колбу на 100 см3 аликвоты раствора b-каротина в этаноле (0,3; 0,5; 1,0; 1,5; 2,0 см3) добавляют растворитель до метки.
Градуировочные растворы других каротиноидов (a-каротина, ликопина, b-криптоксантина, лютеина, зеаксантина) готовят аналогично раствору b-каротина.
Полученные растворы хранят в холодильнике при 2 - 8 °С не более 6 месяцев.
Массовую концентрацию витаминов и каротиноидов в градуировочных растворах (С, мкг/см3) уточняют спектрофотометрическим методом. Для этого определяют оптическую плотность (D) слоя градуировочного раствора толщиной 1 см по отношению к растворителю при длине волны, указанной в таблице, и рассчитывают по формуле:
![]() ,
где
,
где
![]() - коэффициент
светопоглощения, см3×мкг-1×см-1;
- коэффициент
светопоглощения, см3×мкг-1×см-1;
1 - толщина слоя раствора, см;
104 - коэффициент пересчета.
Таблица 11
Условия проведения спектрофотометрических измерений
|
Витамин (каротиноид) |
Растворитель |
Lмакс, нм |
|
Литературный источник |
|
Ретинол |
этанол |
325 |
1832 |
/2/ |
|
a-Токоферол |
этанол |
292 |
75,8 |
/2/ |
|
b-Каротин |
этанол |
453 |
2620 |
/3/ |
|
a-Каротин |
гексан |
445 |
2710 |
/3/ |
|
Ликопин |
гексан |
474 |
3470 |
/3/ |
|
b-Криптоксантин |
гексан |
451 |
2460 |
/3/ |
|
Зеаксантин |
этанол |
452 |
2480 |
/3/ |
|
Лютеин |
этанол |
447 |
2560 |
/3/ |
Градуировка прибора
Градуировка хроматографической системы проводится с целью убедиться, что область определяемых концентраций лежит в пределах линейного участка градуировочного графика, построенного по площадям (высотам) пиков. Она проводится в следующих случаях:
· на этапе освоения методики;
· при смене колонки и/или предколонки;
· при замене стандартного образца;
· в других случаях, когда есть основания полагать, что изменилась эффективность хроматографической системы или чувствительность детектора.
Градуировку хроматографа проводят, строя градуировочный график по серии градуировочных растворов. Аналитический сигнал (площадь, мм2 или высота пика, мм) фиксируют на интеграторе или самописце, соблюдая условия и продолжительность хроматографического разделения и детектирования (табл. 12).
Таблица 12
Условия детектирования витаминов А, Е и каротиноидов
|
Условия детектирования |
|||
|
Витамины (каротиноиды) |
Длина волны детектирования |
Продолжительность, мин |
|
|
Флуориметрический |
Витамин А |
lвозб = 325 нм lэмисс. = 480 нм |
0 - 6 |
|
Витамин Е |
lвозб = 295 нм lэмисс. = 330 нм |
6,1 - 16 |
|
|
Спектрофотометрический |
Каротиноиды |
l = 450 нм |
0 - 30 |
Ориентировочные времена удерживания и последовательность выхода витаминов из хроматографической колонки: ретинол - 4,0 - 4,5 мин, a-токоферол - 10,5 - 11,0 мин (флуориметрическое детектирование), b-каротин > a-каротин > ликопин > b-криптоксантин > лютеин > зеаксантин (спектрофотометрическое детектирование).
Градуировочный график строят в координатах «аналитический сигнал» - «концентрация витамина в градуировочном растворе, мкг/см3». Для каждого анализируемого раствора проводят два параллельных измерения и находят среднее арифметическое. Различие между измеренными значениями аналитических сигналов и времен удерживания не должно превышать 5 % от средних значений. Линейные участки градуировочного графика должны соответствовать всему диапазону определяемых концентраций микронутриентов. Коэффициент градуировочного графика (kгр) определяют как среднее арифметическое значение коэффициентов ki, вычисляемых формуле:
![]() ,
где
,
где
![]() - массовая концентрация микронутриентов в
градуировочном растворе, мкг/см3;
- массовая концентрация микронутриентов в
градуировочном растворе, мкг/см3;
Si - площадь (высота) аналитического сигнала.
Описание хроматографической системы
Подготовку к работе хроматографической системы, состоящей из последовательно соединенных насоса высокого давления, спектрофотометрического и флуориметрического детекторов и интеграторов проводят в соответствии с руководством по эксплуатации.
Используется система для ВЭЖХ следующей конфигурации:
· аналитическая хроматографическая колонка (типа картридж «Элсикарт» фирмы «Элсико», Россия) из нержавеющей стали, длиной 150 мм, внутренним диаметром 4 мм, с обращеннофазным сорбентом, например Нуклеосил 100 С18, зернением 5 мкм или другим, аналогичным по свойствам, с эффективностью по b-каротину не ниже 5000 теоретических тарелок;
· предколонка из нержавеющей стали длиной 50 мм, внутренним диаметром 4 мм с сорбентом типа «Нуклеосил» 100 С18, 7 мкм или аналогичным по свойствам;
· петлевой кран-дозатор (инжектор ввода пробы типа «Реодайн 7125», США) с рабочим объемом петли 50 мм3;
· объемная скорость подачи подвижной фазы - 0,6 см3/мин.
Подготовка пробы к анализу
Подготовка проб к анализу производится непосредственно перед анализом. Взвешивают не менее 10 таблеток (драже, порошкообразное содержимое капсул) и определяют вес одной штуки, затем материал тщательно растирают и перемешивают в фарфоровой ступке. Точную навеску анализируемого материала т, г (0,05 - 0,20 г) помещают в плоскодонную колбу вместимостью 100 см3, добавляют 15 см3 воды и нагревают на водяной бане при температуре от 60 °С до 70 °С при перемешивании в течение 5 мин. Затем прибавляют 30 см3 этилового спирта, 0,1 г аскорбиновой кислоты, 3 см3 50 %-ного раствора гидроокиси калия и нагревают в течение 30 мин на водяной бане с обратным холодильником при температуре кипения смеси. Содержимое колбы быстро охлаждают до комнатной температуры, количественно переносят в делительную воронку и экстрагируют неомыляемые вещества 150 см3 гексана (3 раза по 50 см3) в течение двух минут. Объединенные гексановые экстракты промывают водой по 50 см3 до исчезновения щелочной реакции промывных вод (по универсальной индикаторной бумаге). Промытые гексановые экстракты количественно переносят в колбу объемом V1 (200 см3) и доводят объем раствора до метки тем же растворителем. Аликвоту V2 (2 см3) полученного раствора переносят в мерную пробирку на 10 см3, упаривают в токе азота, сухой остаток растворяют в точно измеренном объеме V3 (1 см3) подвижной фазы.
Измерение концентрации витаминов и каротиноидов в экстракте пробы
Полученный раствор анализируют дважды с помощью хроматографической системы.
Идентификацию пиков проводят сопоставляя времена удерживания и спектральные отношения с временами удерживания и спектральными отношениями растворов соответствующих стандартов (табл. 13).
Таблица 13
Идентификация пиков витаминов и каротиноидов
|
Время удерживания, мин |
Условия детектирования - 1 |
Условия детектирования - 2 |
Соотношения аналитических сигналов (1:2) |
|
|
Ретинол |
4,0 - 4,5 |
lвозб = 325 нм lэмисс. = 480 нм |
lвозб = 340 нм lэмисс. = 95 нм |
1,2 - 1,3 |
|
a-токоферол |
10,5 - 11,0 |
lвозб = 295 нм lэмисс. = 330 нм |
lвозб = 305 нм lэмисс. = 340 нм |
1,9 - 2,0 |
|
b-каротин |
28 - 30 |
450 нм |
470 нм |
1,1 - 1,2 |
|
a-каротин |
24 - 26 |
450 нм |
470 нм |
1,2 - 1,3 |
|
Ликопин |
17 - 18 |
450 нм |
470 нм |
0,7 - 0,8 |
|
b-криптоксантин |
11 - 12 |
450 нм |
480 нм |
1,0 - 1,1 |
|
Лютеин |
5,0 - 5,5 |
450 нм |
480 нм |
1,2 - 1,3 |
|
Зеаксантин |
5,6 - 6,5 |
450 нм |
480 нм |
1,2 - 1,3 |
Концентрацию витаминов и b-каротина в растворе пробы (Спр, мкг/см3) определяют по формуле:
Cnp = kгpSnp, где
Snp - площадь (высота) пика витамина или каротиноида в растворе пробы;
Kгp - коэффициент градуировочного графика.
За окончательный результат определения концентрации микронутриентов в растворе пробы принимают среднее арифметическое результатов параллельных измерений, допускаемое относительное расхождение между которыми не должно превышать 5 % от среднего значения.
Представление результатов
Содержание микронутриентов (X), выраженное в мг на 1 г пищевой добавки, рассчитывают по формуле:
![]() ,
где
,
где
Спр - концентрация витамина или каротиноида в анализируемом растворе (мкг/см3); коэффициент 0,001 учитывает пересчет содержания витаминов и b-каротина в мг.
Минимально детектируемое количество ретинола и a-токоферола - 0,1 мкг/мл, каротиноидов - 0,05 мкг/мл.
Погрешность измерения вычисляется по формуле:
![]() ,
где
,
где
![]() - характеристика погрешности измерений (п. 2), %.
- характеристика погрешности измерений (п. 2), %.
Контроль погрешности результатов измерений
Для контроля погрешности результатов измерений массовой концентрации витаминов А, Е и b-каротина с помощью ВЭЖХ используют метод добавок.
Производят измерение концентрации витаминов и b-каротина в экстракте пробы в соответствии с п. 5.7 (Спр, мкг/см3), а затем с введенной в него добавкой (Сд, мкг/см3), который также анализируют (С¢, мкг/см3). Величину добавки выбирают таким образом, чтобы концентрация микронутриентов в экстракте увеличилась на 50 - 150 %. Для добавки используют градуировочные растворы микронутриентов.
Результаты контроля считают удовлетворительными, если выполняется условие:
![]() ,
где
,
где
Кд - норматив оперативного контроля погрешности, составляющий 15 %;
Сд - расчетное значение величины добавки, мкг/см3.
При превышении норматива оперативного контроля погрешности эксперимент повторяют, выясняют причины, приводящие к неудовлетворительным результатам контроля, и устраняют их.
2. Определение витамина В-6 (пиридоксина) в БАД
Сущность метода
В биологически активных добавках витамин В-6 содержится в основном в виде пиридоксина гидрохлорида, действующим началом которого выступает пиридоксин (витамин В-6). Пиридоксин определяют методом высокоэффективной жидкостной хроматографии (ВЭЖХ) в обращеннофазовом ион-парном варианте в изократическом режиме со спектрофлуориметрическим детектированием. Массовую концентрацию пиридоксина определяют по площади (высоте) пика при соответствующих пиридоксину длинах волн флуориметрического детектирования после введения в хроматографическую систему анализируемых проб и градуировочных растворов.
Характеристика погрешности измерений
Настоящая методика измерений массовой концентрации пиридоксина в биологически активных добавках с вероятностью 0,95 обеспечивает выполнение анализа с погрешностями ±15 % во всем диапазоне измерений.
Описание хроматографической системы
Система для ВЭЖХ с спектрофлуориметрическим детектором с монохроматорами на линии возбуждения и эмиссии (или набором соответствующих фильтров в интервале длин волн от 220 - 650 нм) и аналитической хроматографической колонкой из нержавеющей стали, длиной 150 мм и 250 мм, внутренним диаметром 4 мм и 4,6 мм, с обращеннофазовым сорбентом типа Нуклеосил 100 С18, зернением 5 мкм или другим сорбентом, аналогичным по свойствам, с эффективностью по пиридоксину не ниже 8000 теоретических тарелок. (США) Р7949.
Подготовка к выполнению измерений
Условие выполнения измерений и подготовки проб
Для предотвращения разрушения пиридоксина под действием света подготовку проб и приготовление стандартных растворов пиридоксина проводят в посуде темного стекла, либо, обернув колбочки темной бумагой.
Приготовление растворов
Приготовление раствора соляной кислоты с молярной концентрацией 0,1М.
К дистиллированной воде добавляют 4 см3 концентрированной соляной кислоты (r = 1,19 г/см3), объем доводят до 500 см3.
Раствор фосфорной кислоты с массовой долей 30 %.
41,3 г 85 %-ной фосфорной кислоты (r = 1,698 г/см3) растворяют в дистиллированной воде, доводят объем до 100 см3.
Приготовление 0,03М калий фосфатный буфер, рН 3,0.
4,08 г дигидроортофосфата калия растворяют в 500 см3 дистиллированной воды, доводят рН потенциометрически с помощью раствора фосфорной кислоты с массовой долей 30 % до значения 3,0 и объем доводят до 1000 см3.
Экстрагирующая смесь для гидролиза капсул с содержимым на жировой основе.
40 г винной кислоты растворяют в 800 см3 дистиллированной воды, добавляют 1 г Твина-80, объем доводят до 1000 см3.
Подвижная фаза для ВЭЖХ.
В мерную колбу вместимостью 500 см3 добавляют 100 - 150 см3 0,03М калий фосфатного буфера, 20 см3 ацетонитрила, 326 мг гептилсульфоната натрия и доводят объем до метки фосфатным буфером. Раствор тщательно перемешивают и дегазируют на УЗ-бане в течение 10 мин.
Количественное определение пиридоксина (витамина В-6)
Количественное определение пиридоксина проводят методом абсолютной калибровки, определяя градуировочный коэффициент. Для определения градуировочного коэффициента готовят серию калибровочных растворов (не менее 5) с концентрациями пиридоксина в интервале от 0,01 до 0,200 мкг/см3.
Приготовление градуировочных растворов пиридоксина
Приготовление стандартного раствора пиридоксина гидрохлорида с концентрацией пиридоксина 500 мкг/см3
Точную навеску 61 мг пиридоксина гидрохлорида количественно переносят в мерную колбу вместимостью 100 см3, растворяют в 0,1М растворе соляной кислоты и доводят объем этим же раствором до метки. Содержимое колбы тщательно перемешивают. Концентрацию пиридоксина уточняют спектрофотометрически по коэффициенту молярной экстинкции, равным 7000 M-1×см-1 в 0,1М растворе едкого натра при длине волны 308 нм [2]. Данный раствор можно хранить в холодильнике в течение 3 мес.
Градуировочные растворы пиридоксина готовят непосредственно перед определением. После введения в колбу вместимостью 100 см3 аликвоты 1 см3 приготовленного стандартного раствора объем доводят дистиллированной водой в мерной колбе до 100 см3 и получают раствор с концентрацией 5 мкг/см3. Для приготовления градуировочных растворов в мерные колбы вместимостью 100 см3 помещают аликвоты раствора пиридоксина с концентрацией 5 мкг/см3 (0,2; 0,6; 1,0; 4,0 см3) и доводят объемы до метки дистиллированной водой.
Градуировка прибора
Градуировку хроматографа проводят путем построения градуировочных графиков по серии градуировочных растворов стандартных растворов пиридоксина, приготовленных по п. 5.1. Аналитический сигнал (площадь в мм2 или высота пика в мм) фиксируют на интеграторе или самописце при длине волны возбуждения 290 нм, эмиссии 395 нм. Ориентировочное время удерживания на хроматографической колонке 15,0 - 15,6 мин.
Градуировочный график строят в координатах «аналитический сигнал» - «концентрация пиридоксина в градуировочном растворе, мкг/см3». Для каждого анализируемого раствора проводят два параллельных измерения и находят среднее арифметическое. Различие между измеренными значениями аналитических сигналов и времени удержания не должно превышать 5 % от средних значений. Линейные участки градуировочного графика должны соответствовать всему диапазону определяемых концентраций пиридоксина. Коэффициент градуировочного графика (kгp) определяют как среднее арифметическое значение коэффициентов ki, вычисляемых по формуле:
![]() ,
где
,
где
![]() - массовая концентрация пиридоксина в градуировочном
растворе, мкг/см3,
- массовая концентрация пиридоксина в градуировочном
растворе, мкг/см3,
Si - площадь (высота) аналитического сигнала.
Подготовка проб к анализу
Подготовка проб проводится непосредственно перед анализом.
Таблетки или драже
Взвешивают не менее 10 таблеток (драже) и определяют вес одной (одного) таблетки (драже), затем материал тщательно растирают в фарфоровой ступке. Точную навеску анализируемого материала m, г (0,2 - 1,5 г) помещают в плоскодонную колбу вместимостью 100 см3, добавляют 50 см3 0,1М соляной кислоты, тщательно перемешивают и кипятят на водяной бане в течение 20 мин.
Капсулы с порошкообразным содержимым
3 - 5 желатиновых капсул с порошкообразным содержимым помещают в кислоты и кипятят на водяной бане в течение 20 мин, периодически помешивая.
Капсулы с содержимым на жировой основе
3 - 5 капсул с содержимым на жировой основе помещают в плоскодонную колбу вместимостью 100 см3, добавляют 50 см3 экстрагирующей смеси и кипятят на водяной бане 30 минут, перемешивая через каждые 5 мин. Затем пробы охлаждают до комнатной температуры, помещают на УЗ-баню на 5 мин, доводят экстрагирующей смесью объем до 50 см3 и фильтруют через бумажный фильтр.
При необходимости гидролизаты фильтруют через бумажный фильтр.
Измерение концентрации пиридоксина в экстракте пробы
Перед определением гидролизаты разводят дистиллированной водой в мерных колбах до концентрации, сопоставимой с концентрацией пиридоксина в растворах для построения градуировочного графика (0,03 - 0,05 мкг/см3).
Полученные из экстрактов растворы анализируют дважды с помощью хроматографической системы, вводя на колонку с помощью крана-дозатора (петля 0,1 см3).
Идентификацию пиков проводят, сопоставляя времена удерживания и спекральное отношение с временами удерживния и спектральными отношениями стандартного раствора пиридоксина (табл. 14).
Таблица 14
Идентификация пиков пиридоксина
|
Время удерживания, мин |
Условия детектирования - 1 |
Условия детектирования - 2 |
Соотношение аналитических сигналов (1:2) |
|
|
Пиридоксин |
15,0 - 15,6 |
lвозб = 290 нм lэмисс. = 395 нм |
lвозб = 305 нм lэмисс. = 410 нм |
1,11 |
Концентрацию пиридоксина в растворе пробы (Спр, мкг/см3) определяют по формуле:
![]() ,
где
,
где
Snp - площадь (высота) пика пиридоксина в растворе пробы;
kгp - коэффициент градуировочного графика.
За окончательный результат определения концентрации пиридоксина в растворе пробы принимают среднее арифметическое результатов параллельных измерений, допускаемое относительное расхождение между которыми не должно превышать 5 % от среднего значения.
Обработка результатов
Содержание пиридоксина в 1 таблетке (драже) (мг):
![]() ,
где
,
где
Snp - площадь (высота) пика пиридоксина в пробе;
Kгp - коэффициент градуировочного графика;
V - первоначальный объем гидролизата, см3;
![]() - конечное
разведение гидролизата;
- конечное
разведение гидролизата;
т - навеска таблеток, взятая на анализ;
М - масса таблетки;
1000 - коэффициент пересчета мкг в мг.
Содержание пиридоксина в 1 капсуле (мг):
![]() ,
где
,
где
Snp - площадь (высота) пика пиридоксина в пробе;
kгp - коэффициент градуировочного графика;
V - первоначальный объем гидролизата, см3;
![]() - конечное
разведение гидролизата;
- конечное
разведение гидролизата;
п - количество капсул, взятых на анализ;
1000 - коэффициент пересчета мкг в мг.
Контроль погрешности результатов измерений
Для контроля погрешности результатов измерений массовой концентрации пиридоксина с помощью ВЭЖХ используют метод добавок в гидролизат.
Производят измерение концентрации пиридоксина в гидролизате пробы (Спр, мкг/см3), а затем с введенной в него добавкой (Сд, мкг/см3), который также анализируют (С¢, мкг/см3). Величину добавки выбирают таким образом, чтобы концентрация пиридоксина в гидролизате увеличилась на 50 - 150 %. Для добавки используют калибровочные растворы пиридоксина.
Результаты контроля считают удовлетворительными, если выполняется условие:
![]() ,
где
,
где
Кд - норматив оперативного контроля погрешности (составляет 20 %);
Сд - расчетное значение величины добавки, мкг/см3.
При превышении норматива оперативного контроля погрешности эксперимент повторяют, выясняют причины, приводящие к неудовлетворительным результатам контроля, и устраняют их.
Таблица 15
Метрологические характеристики метода
|
Предел обнаружения пиридоксина |
0,005 мкг в 1 см3 гидролизата |
|
Воспроизводимость, % |
2,0 |
|
Правильность, % |
98,6 |
|
Относительная сходимость, % |
5,0 |
3. Одновременное определение витаминов в-1 и в-2 в бад методом высокоэффективной жидкостной хроматографии
Сущность метода
Витамин В-1 (тиамин) под действием красной кровяной соли превращается в тиохром, который флуоресцирует в щелочной среде. В данном варианте методики происходит постколоночная дериватизация, то есть после отделения тиамина от пробы методом высокоэффективной жидкостной хроматографии (ВЭЖХ) в обращеннофазовом ион-парном варианте в изократическом режиме, а окисляющий агент смешивается с выходящим из колонки элюатом, и образующийся тиохром детектируется флуориметрическим методом [1] при соответствующих длинах волн. Массовую концентрацию рибофлавина определяют по площади (высоте) пика при соответствущих ему длинах волн, так как это соединение способно флуоресцировать в нативном состоянии в растворе.
Характеристика погрешности измерений
Настоящая методика измерений массовой концентрации пиридоксина в биологически активных добавках с вероятностью 0,95 обеспечивает выполнение анализа с погрешностями ±15 % во всем диапазоне измерений.
Описание хроматографической системы
Система для ВЭЖХ со спектрофлуориметрическим детектором с монохроматорами на линии возбуждения и эмиссии (или набором соответствующих фильтров в интервале длин волн от 220 - 650 нм), двухходовым смесителем, в котором происходит реакция образования тиохрома из витамина В-1; и аналитической хроматографической колонкой из нержавеющей стали, длиной 150 мм и 250 мм, внутренним диаметром 4 мм и 4,6 мм, с обращеннофазовым сорбентом типа Нуклеосил 100 С18, зернением 5 мкм или другим сорбентом, аналогичным по свойствам, с эффективностью по тиамину и рибофлавину не ниже 8000 теоретических тарелок.
Подготовка к выполнению измерений
Условие выполнения измерений и подготовки проб
Для предотвращения разрушения рибофлавина и тиамина под действием света подготовку проб и приготовление их стандартных растворов, а также растворов гексацианоферрата (III) калия проводят в посуде темного стекла, либо, обернув колбочки темной бумагой.
Приготовление растворов
Приготовление раствора соляной кислоты с молярной концентрацией 0,1М.
К дистиллированной воде добавляют 4 см3 концентрированной соляной кислоты (r = 1,19 г/см3), объем доводят до 500 см3.
Экстрагирующая смесь для гидролиза капсул с содержимым на жировой основе
40 г винной кислоты растворяют в 800 см3 дистиллированной воды, добавляют 1 г Твина-80, объем доводят до 1000 см3.
3,75 М раствор гидроксида натрия: К 75 г гидроксида натрия добавить 500 см3 дистиллированной воды.
0,04 М раствор гексаноцианоферрата (III) калия: 1,0 г гексаноцианоферрата (III) калия растворить в воде в мерной колбе вместимостью 100 см3 и довести дистиллированной водой до метки.
1,6 103 М щелочной раствор гексаноцианоферрата (III) калия.
2,0 см3 раствора с содержанием гексаноцианоферрата (III) калия 10 г/1000 мл смешать в мерной колбе вместимостью 50 см3 с 3,75 М раствором гидроксида натрия и довести до метки этим же раствором.
Подвижная фаза для ВЭЖХ
В мерную колбу вместимостью 500 см3 добавляют примерно 50 см3 дистиллированной воды, 60 см3 метанола, 100 мг гептилсульфоната натрия, 10 см3 ледяной уксусной кислоты и доводят объем до метки дистиллированной водой. Раствор тщательно перемешивают и дегазируют на УЗ-бане в течение 10 мин.
Количественное определение витаминов В-1 и В-2
Количественное определение витаминов проводят методом абсолютной калибровки, определяя градуировочный коэффициент. Для определения градуировочного коэффициента готовят серию калибровочных растворов (не менее 5) тиамина с концентрациями в интервале от 0,01 до 0,200 мкг/см3, рибофлавина с концентрациями от 0,02 до 0,100 мкг/мл.
Приготовление градуированных растворов тиамина
Приготовление стандартного раствора тиамина гидрохлорида с концентрацией тиамина 1000 мкг/см3.
Точную навеску 62,5 мг тиамина гидрохлорида количественно переносят в мерную колбу вместимостью 50 см3, растворяют в 0,1 М растворе соляной кислоты и доводят объем этим же раствором до метки. Содержимое колбы тщательно перемешивают. Концентрацию пиридоксина уточняют спектрофотометрически по коэффициенту молярной экстинкции, равным 14200 М-1×см-1 в 0,1 М растворе едкого натра при длине волны 247 нм [2]. Данный раствор можно хранить в холодильнике в течение 1 мес.
Градуировочные растворы тиамина готовят непосредственно перед определением. После введения в колбу вместимостью 100 см3 аликвоты 1 см3 приготовленного стандартного раствора объем доводят дистиллированной водой в мерной колбе до 100 см3 и получают раствор с концентрацией 10 мкг/см3. Для приготовления градуировочных растворов в мерные колбы вместимостью 100 см3 помещают аликвоты раствора тиамина с концентрацией 10 мкг/см3 (0,1; 0;2 0,5; 1,0; 2,0 см3) и доводят объемы до метки дистиллированной водой.
Приготовление градуированных растворов рибофлавина
Приготовление стандартного раствора рибофлавина с концентрацией тиамина 100 мкг/см3.
Точную навеску 10,0 мг рибофлавина количественно переносят в мерную колбу вместимостью 100 см3, добавляют примерно 70 см3 воды и нагревают в течение 20 мин до полного растворения, после охлаждения и доводят объем водой до метки. Содержимое колбы тщательно перемешивают. Концентрацию рибофлавина уточняют спектрофотометрически по коэффициенту молярной экстинкции, равным 12500 М-1×см-1 в 0,1М фосфатном буфере (рН = 7,0) при длине волны 255 нм [2]. Данный раствор можно хранить в холодильнике в течение 1 мес.
Градуировочные растворы рибофлавина готовят непосредственно перед определением. После введения в колбу вместимостью 100 см3 аликвоты 1 см3 приготовленного стандартного раствора объем доводят дистиллированной водой в мерной колбе до 50 см3 и получают раствор с концентрацией 2 мкг/см3. Для приготовления градуировочных растворов в мерные колбы вместимостью 100 см3 помещают аликвоты раствора тиамина с концентрацией 10 мкг/см3 (1,0; 2,0; 3,0; 4,0; 5,0 см3) и доводят объемы до метки дистиллированной водой.
Градуировка прибора
Градуировку хроматографа проводят путем построения градуировочных графиков по серии градуировочных растворов стандартных растворов пиридоксина, приготовленных по п. 5.1 - 5.2. Аналитический сигнал (площадь в мм2 или высота пика в мм) фиксируют на интеграторе или самописце при длине волны возбуждения 375 нм, эмиссии 435 нм для тиамина и при длине волны возбуждения 450 нм, эмиссии - 521 нм (для рибофлавина). Ориентировочное время удерживания на хроматографической колонке 15,0 - 15,6 мин.
Градуировочный график строят в координатах «аналитический сигнал» - «концентрация пиридоксина в градуировочном растворе, мкг/см3». Для каждого анализируемого раствора проводят два параллельных измерения и находят среднее арифметическое. Различие между измеренными значениями аналитических сигналов и времени удержания не должно превышать 5 % от средних значений. Линейные участки градуировочного графика должны соответствовать всему диапазону определяемых концентраций витаминов. Коэффициент градуировочного графика (kгp) определяют как среднее арифметическое значение коэффициентов ki, вычисляемых по формуле:
![]() ,
где
,
где
![]() - массовая концентрация витамина в градуировочном
растворе, мкг/см3,
- массовая концентрация витамина в градуировочном
растворе, мкг/см3,
![]() - площадь (высота) аналитического сигнала.
- площадь (высота) аналитического сигнала.
Подготовка проб к анализу
Подготовка проб проводится непосредственно перед анализом. Подготовка проб аналогична таковой при определении пиридоксина, поэтому можно использовать один и тот же гидролизат.
Таблетки или драже
Взвешивают не менее 10 таблеток (драже) и определяют вес одной (одного) таблетки (драже), затем материал тщательно растирают в фарфоровой ступке. Точную навеску анализируемого материала m, г (0,2 - 1,5 г) помещают в плоскодонную колбу вместимостью 100 см3, добавляют 50 см3 0,1М соляной кислоты, тщательно перемешивают и кипятят на водяной бане в течение 20 мин. При необходимости гидролизаты фильтруют через бумажный фильтр.
Капсулы с порошкообразным содержимым
3 - 5 желатиновых капсул с порошкообразным содержимым помещают в кислоты и кипятят на водяной бане в течение 20 минут, периодически помешивая.
Капсулы с содержимым на жировой основе
3 - 5 капсул с содержимым на жировой основе помещают в плоскодонную колбу вместимостью 100 см3, добавляют 50 см3 экстрагирующей смеси и кипятят на водяной бане 30 минут, перемешивая через каждые 5 мин. Затем пробы охлаждают до комнатной температуры, помещают на УЗ-баню на 5 мин, доводят экстрагирующей смесью объем до 50 см3 и фильтруют через бумажный фильтр.
Измерение концентраций тиамина и рибофлавина в экстракте пробы
Перед определением гидролизаты разводят дистиллированной водой в мерных колбах до концентрации, сопоставимой с концентрациями витаминов в растворах для построения градуировочного графика.
Аликвоту фильтрата 0,1 см3 вводили в аналитическую хроматографическую колонку. Витамин В-1 определяли после окисления его в тиохром путем послеколоночной дериватизации 0,06 %-ным раствором калия гексацианоферрата (III) в 3 N гидроокиси калия со скоростью подачи реагента 0,1 см3 в течение 5 мин после ввода пробы. Длина волны возбуждения для тиохрома составляла 375 нм, длина волны флуоресценции - 435 нм. Время удерживания витамина В-1 - 4,0 - 4,5 мин. После 6-й минуты длины волн автоматически переключались для определения витамина В-2. Длина волны возбуждения для рибофлавина (витамина В-2) составляла 450 нм, длина волны флуоресценции - 521 нм. Время удерживания витамина В-2 составляло 10,0 - 10,3 мин. Идентификацию пиков проводили, сопоставляя времена удерживания с временами удерживания растворов соответствующих стандартов.
Идентификацию пиков проводят, сопоставляя времена удерживания и спектральное отношение с временами удерживания и спектральными отношениями стандартного раствора пиридоксина (табл. 16).
Таблица 16
Идентификация пиков тиамина рибофлавина
|
Время удерживания, мин |
Условия детектирования - 1 |
Условия детектирования - 2 |
Соотношение аналитических сигналов (1:2) |
|
|
Рибофлавин |
10,0 - 10,3 |
lвозб = 450 нм lэмисс. = 521 нм |
lвозб = 465 нм lэмисс. = 535 нм |
1,12 |
|
Тиамин |
4,0 - 4,5 |
lвозб = 375 нм lэмисс. = 435 нм |
lвозб = 395 нм lэмисс. = 450 нм |
1,01 |
Концентрацию витаминов в растворе пробы (Спр, мкг/см3) определяют по формуле:
Cnp = kгp×Snp, где
Snp - площадь (высота) пика витаминов в растворе пробы;
Kгp - коэффициент градуировочног графика.
За окончательный результат определения концентрации пиридоксина в растворе пробы принимают среднее арифметическое результатов параллельных измерений, допускаемое относительное расхождение между которыми не должно превышать 5 % от среднего значения.
Обработка результатов
Содержание тиамина (рибофлавина) в 1 таблетке (драже) (мг):
![]() ,
где
,
где
Snp - площадь (высота) пика витамина в пробе;
kгp - коэффициент градуировочного графика;
V - первоначальный объем гидролизата, см3;
kразв - конечное разведение гидролизата;
т - навеска таблеток, взятая на анализ;
М - масса таблетки;
1000 - коэффициент пересчета мкг в мг.
Содержание тиамина (рибофлавина) в 1 капсуле (мг):
![]() ,
где
,
где
Snp - площадь (высота) пика пиридоксина в пробе;
kгp - коэффициент градуировочного графика;
V - первоначальный объем гидролизата, см3;
kразв - конечное разведение гидролизата;
п - количество капсул, взятых на анализ;
1000 - коэффициент пересчета мкг в мг.
Контроль погрешности результатов измерений
Для контроля погрешности результатов измерений массовой концентрации пиридоксина с помощью ВЭЖХ используют метод добавок в гидролизат.
Производят измерение концентрации тиамина или рибофлавина в гидролизате пробы (Спр, мкг/см3), а затем с введенной в него добавкой (Сд, мкг/см3), который также анализируют (С¢, мкг/см3). Величину добавки выбирают таким образом, чтобы концентрация данного витамина в гидролизате увеличилась на 50 - 150 %. Для добавки используют калибровочные растворы.
Результаты контроля считают удовлетворительными, если выполняется условие:
![]() ,
где
,
где
Кд - норматив оперативного контроля погрешности (составляет 20 %);
Сд - расчетное значение величины добавки, мкг/см3.
При превышении норматива оперативного контроля погрешности эксперимент повторяют, выясняют причины, приводящие к неудовлетворительным результатам контроля, и устраняют их.
Таблица 17
Метрологические характеристики метода
|
Характеристика |
Тиамин |
Рибофлавин |
|
Предел обнаружения |
0,005 мкг в 1 мл гидролизата |
0,006 мкг в 1 мл гидролизата |
|
Воспроизводимось, % |
3 |
3 |
|
Правильность, % |
98,1 |
98,7 |
4. Флуориметрический метод определения рибофлавина (витамин В2) титрованием рибофлавинсвязывающим апобелком
Принцип метода основан на способности рибофлавинсвязывающего апобелка при связывании с рибофлавином полностью тушить его флуоресценцию.
Реактивы и их приготовление
0,1 н. соляная кислота (НСl).
К дистиллированной воде добавляют 0,8 см3 концентрированной (r = 1,19 г/см3) соляной кислоты по ГОСТ 3118-77, объем доводят до 100 см3.
Стандартный раствор рибофлавина (С17H20N4O6) концентрацией 100 мкг/см3.
5 мг рибофлавина растворяют при нагревании на водяной бане при 70 °С в 45 см3 0,1 н. соляной кислоты (раствор 1). После охлаждения объем доводят до 50 см3. Концентрацию рибофлавина уточняют спектрофотометрически по коэффициенту молярной экмтинкции (прилож. 3). Раствор можно хранить в холодильнике в течение 6 мес.
Рабочий стандартный раствор (концентрация рибофлавина 2 мкг/см3).
Перед определением к 1 см3 стандартного раствора добавляют дистиллированную воду в мерной колбе до объема 50 см3.
4 М фосфат калия однозамещенный (К2НРО4).
54,4 г К2НРО4 растворяют в дистиллированной воде, объем доводят до 100 см3.
Рибофлавинсвязывающий апобелок (РбСБ) со связывающей способностью 5 - 15 мкг рибофлавина на 1 мг белка.
Можно использовать коммерческий препарат (Sigma, США, R 8628) или выделить его из белка куриного яйца по методу, изложенному в прилож. 3.
Средства измерений
Спектрофлуориметр F-2000 (Hitachi, Япония) или аналогичный по оптическим характеристикам.
Условия измерения
Интенсивность флуоресценции измеряют в кварцевой кювете объемом 3 см3 с длиной оптического пути 1 см при длине волны возбуждающего света lвозб = 465 нм, испускаемого lфл = 525 нм.
Приготовление кислотного гидролизата
3 таблетки (драже) или содержимое 3 капсул растирают и тщательно перемешивают в ступке. При расчете массы навески исходят из того, чтобы в 1 см3 гидролизата содержалось не более 5 мкг рибофлавина. К навеске (100 - 500 мг в зависимости от содержания витамина) добавляют 100 см3 0,1 н. НСl и помещают на кипящую водяную баню на 40 мин. Затем пробы охлаждают до комнатной температуры, доводят объем дистиллированной водой до 100 см3 и при необходимости фильтруют.
В случае необходимости разводят гидролизат до концентрации рибофлавина около 0,5 мкг/см3 или уменьшают количество гидролизата, взятого на анализ, с таким расчетом, чтобы интенсивность флуоресценции опытной пробы (о - к) была сопоставима с интенсивностью флуоресценции добавленного в пробу стандарта (с - о).
Ход определения
К 0,1 см3 кислотного гидролизата добавляют равный объем 4М К2НРО4 и дистиллированную воду до 3 см3, измеряют флуоресценцию (о), добавляют 0,03 см3 стандартного раствора рибофлавина концентрацией 2 мкг/см3. После перемешивания повторяют измерение (с). Затем добавляют по 0,03 см3 раствора РбСБ, перемешивают, измеряют флуоресценцию после каждой добавки белка. Добавление белка продолжают до тех пор, пока флуоресценция после двух добавок перестает снижаться (к). Разница интенсивности флуоресценции (о - к) пропорциональна количеству рибофлавина в гидролизате. Необходимое количество РбСБ подбирают эмпирически.
Обработка результатов
Содержание рибофлавина в 1 таблетке (мг):
![]() ,
где
,
где
0,06 - количество рибофлавина, внесенное в кювету, мкг;
V - исходный объем гидролизата, см3;
М - масса таблетки БАД, г;
V1 - объем гидролизата, внесенного в пробу, см3;
т - навеска измельченных таблеток, взятая на анализ, г;
1000 - перевод мкг в мг.
Таблица 18
Метрологические характеристики метода
|
Предел обнаружения |
0,03 мкг в 1 мл гидролизата |
|
Воспроизводимость, % |
6,4 |
|
Правильность, % |
97,1 |
|
Относительная сходимость, % |
18 |
5. Метод определения аскорбиновой кислоты (витамин С)
Определение витамина С в биологически активных добавках к пище основано на определении непосредственно аскорбиновой кислоты (АК) без учета окисленной формы витамина С - дегидроаскорбиновой кислоты (ДАК).
Для целей рутинного анализа наиболее легко и доступно определение методом визуального титрования с использованием количественного окисления АК раствором 2,6-дихлорфенолиндофенолята натрия. Однако этот метод применим только для объектов исследования, дающих светлоокрашенные экстракты. В других случаях используют метод потенциометрического титрования, спектрофотометрические и флуорометрический методы анализа, которые применимы для любых объектов исследования.
Аппаратура, материалы и реактивы
Спектрофотометр с диапазоном измерения от 220 до 1100 нм или фотоэлектроколориметр с диапазоном измерения от 364 до 2980 нм с диапазоном измерения коэффициента пропускания от 100 до 1 %.
Флуориметр типа «Квант» с набором светофильтров или спектрофлюориметр.
Весы лабораторные общего назначения 2-го класса точности с наибольшим пределом взвешивания 200 г по ГОСТ 24104.
Весы лабораторные общего назначения 4-го класса точности с наибольшим пределом взвешивания 500 г по ГОСТ 24104.
Секундомер 2-го класса или часы 2-го класса.
РН-метр-милливольтметр лабораторный для измерения рН и окислительно-восстановительных потенциалов с диапазонами от минус 1 до плюс 14 мВ и погрешностью не более ±0,05 при измерении рН и диапазонами от минус 100 до плюс 1400 мВ и погрешностью не более ±5 мВ при измерении окислительно-восстановительных потенциалов. При измерении окислительно-восстановительных потенциалов используют электроды: измерительный - платиновый, вспомогательный - хлорсеребряный.
Термостат с поддержанием заданного режима от 0 до 60 °С с погрешностью ±0,8 °С.
Термометр жидкостный с диапазоном измерения от 0 до 100 °С с ценой деления шкалы 1 °С.
Мешалка магнитная с плавным регулированием частоты вращения.
Воронки стеклянные лабораторные типа В по ГОСТ 25336.
Колбы мерные лабораторные стеклянные номинальной вместимостью 50, 100, 500, 1000 см3 по ГОСТ 1770.
Колбы типа Кн исполнения 2 номинальной вместимостью 50, 100, 250, 750 см3 по ГОСТ 25336.
Микробюретка по ГОСТ 20292 с ценой деления не более 0,01 см3.
Палочки стеклянные.
Пипетки мерные лабораторные стеклянные по ГОСТ 20292 номинальной вместимостью 1, 2, 5, 10 см3 ценой деления шкалы 0,1 см3.
Стаканы типа В исполнения 1 номинальной вместимостью 50, 100, 400, 1000 см3 по ГОСТ 25336.
Ступка фарфоровая с пестиком по ГОСТ 9147.
Цилиндры мерные лабораторные стеклянные вместимостью 100, 250 см3 по ГОСТ 1770.
Бумага фильтровальная лабораторная по ГОСТ 12026.
Бумага масштабно-координатная (миллиметровая) по ГОСТ 334.
Вода дистиллированная по ГОСТ 6709.
Ацетон по ГОСТ 2603.
Кислота азотная по ГОСТ 4461 плотностью 1,41 г/см3, раствор с объемной долей 25 %.
Кислота аскорбиновая по ГФ XI изд., растворы массовых концентраций 1,0 и 0,1 г/дм3.
Кислота борная, ч.
Кислота серная о.с.ч. или ч.д.а. (предпочтительней по Савалю).
Кислота соляная плотностью 1,19 г/см3, х.ч., раствор с массовой долей 2 %.
Кислота трихлоруксусная, х.ч., раствор с массовой долей 3 %.
Кислота фосфорная (мета), ч. по ГОСТ 841, растворы с массовой долей 3 и 6 %.
Кислота уксусная ледяная, ч.д.а. или х.ч. по ГОСТ 61, раствор с массовой долей 3 %.
Кислота хлорная, раствор молярной концентрации 0,1 моль/дм3. Готовят перед обработкой электродов.
Кислота щавелевая, раствор с массовой долей 2 %.
Ксилол.
Натрий 2,6-дихлорфенолиндофенолят, х.ч., Serva
Натрий уксуснокислый, 3-водный, ч.д.а. по ГОСТ 199.
О-Фенилендиамин, раствор 0,2 г/дм3.
Формальдегид, 36 - 40 %-ный раствор.
Уголь активированный.
L-цистеин солянокислый или L-цистеин, 50 мг в 1 см3 2 %-ного раствора соляной кислоты.
Этилендиаминтетраацетат (ЭДТА) или трилон Б (динатриевая соль этилендиамина-N, N¢, N¢-тетрауксусной кислоты, 2-водная, ч.д.а.).
Эфир уксусной кислоты бутиловый (бутилацетат), х.ч.
Допускается применение других средств измерений с метрологическими характеристиками и оборудования с техническими характеристиками не хуже, а также реактивов и материалов по качеству не ниже вышеуказанных.
Хранение растворов по ГОСТ 4212.
Методы отбора проб
Подготовка средней пробы к анализу
Подготовка проб к анализу производится непосредственно перед анализом. Выделение витамина необходимо осуществлять возможно быстрее, без использования повышенной температуры, не на ярком свету и при минимальном контакте образца с кислородом воздуха.
Дражированная форма
Из средней пробы драже отбирают 10 - 15 шт., взвешивают их и определяют вес одной штуки, затем тщательно растирают и перемешивают в фарфоровой ступке.
Таблетированная форма
Из средней пробы таблеток отбирают 3 - 7 шт., взвешивают их и определяют вес одной штуки, затем тщательно растирают и перемешивают в фарфоровой ступке.
Капсулированная форма
Из средней пробы капсул с порошкообразным содержимым отбирают 3 - 7 шт., взвешивают их и определяют вес одной штуки за вычетом массы оболочки, затем содержимое капсул тщательно растирают и перемешивают в фарфоровой ступке.
Из средней пробы «жировых» капсул отбирают 3 - 7 шт., взвешивают их, затем осторожно вскрывают, содержимое капсул тщательно перемешивают. Оболочки капсул отмывают органическим растворителем (ацетоном, гексаном и др.), высушивают и взвешивают. Затем высчитывают массу содержимого одной капсулы.
Кристаллические витаминные препараты (субстанции и премиксы)
Пробу витаминных препаратов отбирают в количестве 5 - 10 г и тщательно перемешивают.
Порошкообразная форма
Среднюю пробу порошкообразных веществ (сухие напитки, сухие молочные смеси и каши для детского питания и т.д.) отбирают в количестве 25 - 50 г. Перед взятием пробы порошок тщательно перемешивают.
Примечание. При исследовании равномерности перемешивания сыпучих продуктов, расфасованных в порционные упаковки, целесообразно при проведении анализа использовать порционную упаковку исследуемого образца целиком.
Жидкие витаминные препараты и обогащенные продукты
Жидкие витаминные препараты и обогащенные продукты в количестве 50 - 100 см3 осторожно, избегая аэрации, перемешивают путем многократного переворачивания склянки, содержащей пробу.
Витаминизированные кондитерские изделия (конфеты, вафли, батончики, бисквиты и др.)
Пробу штучных изделий берут в количестве не менее 100 - 200 г (в зависимости от содержания витамина), взвешивают, определяют вес единицы изделия, а затем подвергают размельчению и растиранию в ступке.
Подготовка аналитической пробы
Из измельченной и перемешанной пробы берут навеску не менее 0,5 - 1 г, взвешенную с погрешностью ±0,0001 г. При расчете массы необходимой навески можно пользоваться следующей формулой:
![]() ,
где
,
где
т - навеска исследуемого образца, г
![]() - общий объем
экстракта, мл
- общий объем
экстракта, мл
Vnp - объем экстракта, взятого на титрование, мл
![]() - заявленное
содержание АК в исследуемом образце, мг/100 г.
- заявленное
содержание АК в исследуемом образце, мг/100 г.
Навески жидких проб (витаминизированные соки, напитки и т.п.) отбирают пипеткой. Навески проб густой консистенции (сиропы, концентраты и пр.), плохо стекающие из пипетки, берут весовым путем подобно твердым продуктам. Затем в этих пробах определяют по общим правилам плотность для пересчета содержания витамина на единицу объема.
5.1 Определение витамина С в образцах, дающих неокрашенные или слабоокрашенные экстракты
Сущность метода
Определение витамина С в образцах, дающих неокрашенные или слабоокрашенные экстракты основано на экстрагировании аскорбиновой кислоты или ее солей раствором кислоты (соляной, щавелевой, трихлоруксусной, метафосфорной или смесью уксусной и метафосфорной) с последующим визуальным титрованием раствором 2,6-дихлорфенолиндофенолятом натрия до установления светло-розовой окраски.
Подготовка к выполнению измерения (испытанию)
Выбор экстрагирующего раствора
Таблица 19
Экстрагирующие агенты
|
Экстрагирующий агент |
Область применения |
|
1 |
2 |
|
2 % соляная кислота |
Дражированные, таблетированные, капсулированные, кристаллические формы БАД, обогащенные продукты питания, без длительного хранения экстрактов. Исключая продукты с высоким содержанием белка |
|
2 % щавелевая кислота |
То же, но возможно хранение экстрактов в течение 4 - 5 ч |
|
3 % трихлоруксусная кислота |
Для всех видов БАД и обогащенных продуктов питания, включая продукты с высоким содержанием белка |
|
Смесь уксусной и метафосфорной кислот |
То же |
|
6 % метафосфорная кислота |
То же, возможно хранение экстракта в течение 4 - 5 ч |
При выборе экстрагирующего агента также следует учитывать влияние мешающих определению компонентов исследуемой матрицы (табл. 20).
Таблица 20
Компоненты, мешающие определению АК
|
Устранение влияния мешающих компонентов |
|
|
Диоксид серы (SО2) |
В экстракт
добавляют объем ацетона, равный |
|
Наличие ионов двухвалентных металлов (Fe2+, Cu2+, Sn2+ и др.) |
Экстрагирование проводят, смешивая равные объемы 0,18 % раствора ЭДТА и 6 % раствора метафосфорной кислоты (3 % раствора ТХУ). |
|
Высокое содержание жира |
Использование в качестве экстрагирующего агента смеси 6 % раствора метафосфорной кислоты и ацетона в соотношении 1:3 |
|
Жирорастворимые витамины в составе «жировых» капсул |
Аналитическую пробу исследуемого БАД перед проведение экстракции следует предварительно растворить в небольшом объеме органического растворителя (эфир, хлороформ, гексан) |
|
Восстанавливающие сахара (редуктоны) |
Обработка экстракта формалином при рН 3,5 приводит к полной дезактивации АК, при рН 0 - редуктонов. |
|
Сульфгидрильные соединения (цистеин, глутатион восстановленный, белки, танин и т.п.) |
Обработка экстракта 5 % раствором уксуснокислого свинца. |
|
Наличие в исследуемом образце большого количества зерновых |
В процессе экстрагирования следует добавить к объекту исследования 2 - 5 см3 метанола. |
Приготовление экстрагирующего раствора
Приготовление водного раствора соляной кислоты массовой концентрации 20 г/дм3 (2 %)
47,5 см3 36 %-ной соляной кислоты доводят до метки в 1000 см3 мерной колбе дистиллированной водой.
Приготовление водного раствора трихлоруксусной кислоты массовой концентрации 30 г/дм3 (3 %).
30 г кристаллической трихлоруксусной кислоты доводят до метки в 1000 см3 мерной колбе дистиллированной водой.
Приготовление водного раствора метафосфорной кислоты массовой концентрации 60 г/дм3
Взвешивают 60,00 г растертой в ступке метафосфорной кислоты на весах 4-го класса точности, растворяют без нагревания в 100 - 200 см3 дистиллированной воды в мерной колбе вместимостью 1000 см3, доводят дистиллированной водой до метки, перемешивают и фильтруют. Раствор хранят при (6±2) °С не более 7 сут.
Приготовление водного раствора метафосфорной кислоты массовой концентрации 30 г/дм3
Необходимый для измерения объем водного раствора метафосфорной кислоты массовой концентрации 60 г/дм3 смешивают с дистиллированной водой в соотношения 1:1. Раствор готовят непосредственно перед применением.
Приготовление водного раствора трилона Б (этилендиаминтетраацетат натрия) молярной концентрации с = 0,05 моль/дм3
Взвешивают 1,8 г трилона Б на весах 2 класса точности и растворяют в 100 - 300 см3 дистиллированной воды в мерной колбе вместимостью 1000 см3, доливая дистиллированную воду до метки.
Приготовление смеси водного раствора трилона Б (этилендиаминтетраацетат натрия) в молярной концентрации с = 0,05 моль/дм3 и раствора метафосфорной кислоты массовой концентрации 60 г/дм3 или раствора трихлоруксусной кислоты массовой концентрации 30 г/дм3 (раствор осадителя)
Отмеряют цилиндром 300 см3 водного раствора метафосфорной кислоты массовой концентрации 60 г/дм3 (или водного раствора трихлоруксусной кислоты массовом концентрации 30 г/дм3) и 300 см3 раствора трилона Б (этилендиаминтетраацетат натрия) молярной концентрации с = 0,05 моль/дм3 в коническую колбу объемом 750 см3 и перемешивают. Раствор готовят непосредственно перед применением.
Приготовление смеси уксусной и метафосфорной кислот
К раствору 15 г метафосфорной кислоты в 200 см3 дистиллированной воды добавляют 40 см3 ледяной уксусной кислоты и доводят дистиллированной водой до 500 см3. Хранят в склянке с притертой пробкой в холодильнике в течение 7 дней.
Приготовление стандартного раствора АК
Растворяют 0,1000 г (точная навеска) аскорбиновой кислоты, отвечающей требованиям ГФ XI, в мерной колбе емкостью 100 см3 в выбранном экстрагирующем растворе и доводят тем же раствором до метки, 10 см3 полученного раствора АК помещают в мерную колбу на 100 см3 и доводят до метки раствором, используемым в качестве экстрагирующего. Раствор готовят непосредственно перед применением.
Приготовление раствора 2,6-дихлорфенолиндофенолята натрия (раствор красителя) и определение его титра
Взвешивают 0,100 г 2,6-дихлорфенолиндофенолята натрия на весах 2 класса точности, растворяют, тщательно перемешивая, в 250 см3 свежекипяченой дистиллированной воды температурой (90±5) °С содержащей около 50 мг бикарбоната натрия. После охлаждения раствора до (20±5) °С, доводят до метки дистиллированной водой в мерной колбе вместимостью 500 см3, затем фильтруют через складчатый фильтр в склянку из темного стекла. Раствор хранят при (6±2) °С не более 1 месяца.
Определение титра
К 1 мл стандартного раствора АК добавляют 9 см3 выбранного экстрагирующего раствора и титруют раствором 2,6-дихлорфенолиндофенолята натрия до розового окрашивания, не исчезающего в течение 15 - 20 с (Vm). Таким же образом титруют 10 см3 экстрагирующего раствора (Vк). Поправку к титру раствора (Т), в мг АК, эквивалентных 1 мл раствора 2,6-дихлорфенолиндофенолята натрия, вычисляют по формуле:
![]() ,
где
,
где
К - количество АК в 1 мл стандартного раствора, мг.
Приготовление раствора ацетатного буфера с рН 4,0
Растворяют 30 г безводного уксуснокислого натрия в 70 см3 дистиллированной воды и, добавляя ледяную уксусную кислоту (около 100 см3), потенциометрически доводят рН до 4,0.
Приготовление раствора ацетатного буфера с рН 5,0
Растворяют 30 г безводного уксуснокислого натрия в 70 см3 дистиллированной воды и, добавляя ледяную уксусную кислоту (около 50 см3), потенциометрически доводят рН до 5,0.
Приготовление 1,0 М раствора соляной кислоты
17,2 мл 36 % соляной кислоты разбавляют дистиллированной водой в мерной колбе емкостью на 200 см3 и доводят до метки дистиллированной водой.
Приготовление 1,0 М раствора ацетата натрия
82,0 г безводного ацетата натрия растворяют в мерной колбе емкостью 1000 см3 дистиллированной водой и доводят до метки.
Приготовление солянокислого буфера с рН 5,2
К 40 см3 1,0 М раствора соляной кислоты приливают 200 см3 1,0 М раствора ацетата натрия и доводят до метки дистиллированной водой в мерной колбе емкостью 1000 см3.
Проведение испытания
Экстрагирование
Для приготовления экстракта рассчитанную массу навески взвешивают с погрешностью ±0,0001 г. Затем гомогенизируют ее с небольшим количеством выбранного экстрагирующего агента. После чего полученный гомогенат количественно переносят в мерную колбу или мерный цилиндр объемом Vобщ, доводят до метки экстрагирующим агентом, тщательно перемешивают, настаивают 5 - 10 мин и фильтруют через складчатый фильтр или центрифугируют. Для жидких пищевых добавок необходимое количество материала отмеривают пипеткой и доводят до метки экстрагирующим агентом в соответствующей мерной колбе.
Визуальное титрование
Для определения АК 1 - 10 мл экстракта с общим содержанием АК около 0,1 мг вносят пипеткой в коническую колбу вместимостью 50 или 100 см3, доводят объем экстрагентом до 10 см3 и титруют раствором 2,6-дихлор-фенолиндофенолята натрия до слабо-розового окрашивания, не исчезающего в течение 15 - 20 с. Аналогично титруют равный объем экстрагирующего агента (контроль на реактивы)*.
Обработка результатов измерения
Содержание АК (X), выраженное в мг на 1 г пищевой добавки вычисляют по формуле:
![]() ,
где
,
где
Т - титр раствора 2,6-дихлорфенолиндофенолята натрия, мг/см3;
Vm - объем раствора 2,6-дихлорфенолиндофенолята натрия, пошедший на титрование исследуемого раствора, см3;
Vк - объем раствора 2,6-дихлорфенолиндофенолята натрия, пошедший на контрольное испытание, см3;
остальные обозначения см. выше.
Для пищевых добавок, содержащих редуцирующие примеси вместо контроля на реактивы проводят контрольное испытание на присутствие этих примесей. Для этого в колбу помещают такой же объем экстракта, как для определения АК, прибавляют равный ему объем ацетатного буферного раствора с рН 4,0, 36 - 40 %-ный раствор формальдегида в объеме, равном половине объема буферного раствора, перемешивают и выдерживают в течение 10 мин, закрыв предварительно колбу пробкой. Затем содержимое титруют раствором 2,6-дихлорфенолиндофенолята натрия.
За окончательный результат принимают среднее арифметическое результатов двух параллельных определений и не менее, чем с двумя различными навесками, различающимися не более, чем на 10 %. Чувствительность визуального титрования составляет 5 мкг/см3.
5.2 Определение витамина С в образцах, дающих окрашенные экстракты
Флуориметрический метод определения
Этот метод рассмотрен в «Руководстве по методам анализа качества и безопасности пищевых продуктов», 1998 г.
Метод индофенол-ксилолъной экстракции
Этот метод рассмотрен в сборнике научных трудов «Теоретические и клинические аспекты науки о питании» т. VIII, 1987, с. 178 - 180.
Колориметрический анализ АК в водной среде
Приготовление экстракта и реактивов см. в п. 5.2.
Проведение анализа. Готовят растворы в следующей последовательности (табл. 21).
Таблица 21
Последовательность добавления реагентов в колориметрическом методе
|
Реагент |
№ раствора (колбы) |
|||||
|
1 |
1* |
2 |
3 |
3* |
||
|
контроль |
проба сравнения |
стандарт |
экстракт |
проба сравнения экстракта |
||
|
1 |
Ацетатный буфер рН 5,0* |
2,5 мл |
2,5 мл |
2,5 мл |
2,5 мл |
2,5 мл |
|
2 |
Экстракт |
- |
- |
- |
5 мл |
5 мл |
|
3 |
Стандартный раствор АК |
- |
- |
0,5 мл |
- |
- |
|
4 |
Экстрагирующий агент (3 % НРО3 или 2 % НСl) |
5 мл |
5 мл |
4,5 мл |
- |
- |
|
5 |
Дистиллированная вода |
- |
2 мл |
- |
- |
2 мл |
|
6 |
Фенолят |
2 мл |
- |
2 мл |
2 мл |
- |
При экстракции 2 % НСl на анализ берут по 5 см3 ацетатного буфера.
При большой концентрации АК или интенсивной окраске экстракта берут меньше 5 см3 экстракта, добавляя в растворы № 3 и 3* количество экстрагирующего агента равное 5 см3 минус объем пробы. Измеряют поглощение фенолята (избытка) в пробах № 1 - 3 при l = 540 нм.
Содержание АК (X), выраженное в мг на 100 г исследуемого образца рассчитывают по формуле:
![]() ,
где
,
где
К - количество АК в 0,5 см3 стандартного раствора, взятого на определение, мг;
![]() - поглощение
фенолята в контрольном растворе (№ 1) против поглощения фенолята в растворе
стандарта (№ 2);
- поглощение
фенолята в контрольном растворе (№ 1) против поглощения фенолята в растворе
стандарта (№ 2);
![]() - поглощение
фенолята в контрольном растворе № 1 (кювета сравнения - раствор № 1*);
- поглощение
фенолята в контрольном растворе № 1 (кювета сравнения - раствор № 1*);
![]() - поглощение
фенолята в растворе экстракта № 3 (кювета сравнения - раствор № 3*);
- поглощение
фенолята в растворе экстракта № 3 (кювета сравнения - раствор № 3*);
остальные обозначения приведены выше.
За окончательный результат принимают среднее арифметическое двух определений
5.3. Спектрофотометрическое определение витамина С в напитках
Проведение анализа
После дегазации и, если необходимо, фильтрования исследуемых напитков готовят три раствора, добавляя реагенты в следующей последовательности (табл. 22).
Таблица 22
Последовательность добавления реагентов при спектрофотометрическом определении витамина С
|
Раствор №, см3 |
|||
|
1 |
2 |
3 |
|
|
контроль на реактивы |
стандарт |
исследуемый раствор |
|
|
Солянокислый буфер (рН 5,2) |
5,0 |
4,5 |
4,5 |
|
Напиток |
- |
- |
0,5 |
|
Стандартный раствор аскорбиновой кислоты |
- |
0,5 |
- |
|
Раствор 2,6-дихлорфенолиндофенолята натрия |
1,0 |
1,0 |
1,0 |
Измеряют поглощение фенолята (избытка) в пробах № 2 - 3 при l = 540 нм.
Содержание АК (X), выраженное в мг на 1 л напитка рассчитывают по формуле:
![]() ,
где
,
где
K - количество АК в 0,5 см3 стандартного раствора, взятого на определение, мг;
![]() - величина
поглощения исследуемого раствора № 3 против поглощения фенолята в контроле на
реактивы;
- величина
поглощения исследуемого раствора № 3 против поглощения фенолята в контроле на
реактивы;
![]() - общий объем
исследуемого напитка, обычно его принимают равным 1000 см3;
- общий объем
исследуемого напитка, обычно его принимают равным 1000 см3;
![]() - величина
поглощения стандартного раствора № 2 против поглощения фенолята в контроле на
реактивы;
- величина
поглощения стандартного раствора № 2 против поглощения фенолята в контроле на
реактивы;
Vnp - объем исследуемого напитка, взятый для спектрофотометрирования, в данном случае - 0,5 см3.
За окончательный результат определения принимают среднее арифметическое двух определений. Расхождение между двумя параллельными определениями не должно превышать 3 % от среднего арифметического.
Чувствительность - 3 мкг АК в пробе.
Для одной партии буфера и одного и того же раствора 2,6-дихлорфенолиндофенолята натрия величину (K/D1-2), характеризующую качество реактива Тильманса, можно определять один раз в течение одной - двух недель, считая ее постоянной.
5.4. Потенциометрическое титрование
Этот метод описан в «Руководство по методам анализа качества и безопасности пищевых продуктов», 1998.
Приложение 1
Некоторые физико-химические свойства витаминов
Аскорбиновая кислота
Белые кристаллы. Устойчива в твердом состоянии. Водные растворы имеют рН 3. Водные растворы устойчивы только в отсутствие кислорода, на воздухе устойчивы при рН 5 - 6, очень неустойчивы при щелочных рН. Окисление катализируют Сu, Fe. Хорошо растворима в воде - 33,3 г в 100 см3 при комнатной температуре.
Тиаминхлорид гидрохлорид
Белые кристаллы. Гигроскопичен. Устойчив в твердом состоянии на воздухе. Водные растворы при рН > 5 не устойчивы к нагреванию, при рН > 7,0 не устойчивы при комнатной температуре. Хорошо растворим в воде - 100 г в 100 см3 при комнатной температуре.
Рибофлавин
Желтые кристаллы. Сухое твердое вещество устойчиво к рассеянному свету. Чрезвычайно фотолабилен в растворах, особенно щелочных. Нейтральные и кислые водные растворы устойчивы в темноте, за 1 мес. разлагается 3 % при 27 °С и рН 6,0. Быстро разлагается в щелочных растворах. Мало растворим в воде - 0,01 г в 100 см3 при 25 °С и 0,23 г в 100 см3 при 100 °С. Растворимость в кислых растворах: в 3 %-ной соляной кислоте при 15 °С - 0,026 %.
Пиридоксингидрохлорид
Белые кристаллы. Устойчив в твердом состоянии на свету. Нейтральные и щелочные растворы фотолабильны. Хорошо растворим в воде - 22 г в 100 см3 при комнатной температуре.
a-токоферол
Светло-желтые масла. Хорошо растворим в этаноле. Медленно окисляется на воздухе, быстро - в присутствии щелочи или при нагревании. Устойчив к действию кислот (до 100 °С). Постепенно темнеет на свету, чувствителен к УФ-свету.
a-токоферилацетат
Желтые кристаллы. Эфиры (ацетат и др.) гораздо более устойчивы к свету и кислороду.
Ретинол
Желтые кристаллы. Легко окисляется на воздухе. Разрушается под действием УФ-света. Устойчив в щелочной среде, неустойчив - в кислой. Хорошо растворим в этаноле.
b-каротин
Темно-фиолетовые кристаллы. Окисляется на воздухе, фотолабилен. Растворим в этаноле.
Приложение 2
Определение концентрации растворов витаминов, используемых в качестве стандартов
Таблица 23
Коэффициенты молярной экстинкции витаминов для уточнения концентрации стандартных растворов
|
Витамин |
Молекулярная масса |
Условия измерения |
l, нм |
Е1 см |
|
Аскорбиновая кислота |
176 |
вода |
265 |
7000 |
|
кислота |
245 |
7500 |
||
|
Тиамин |
265 |
0,1 М трис-НСl рН 7,5 |
267 |
8300 |
|
Рибофлавин |
376 |
0,1 М фосфат рН 7,0 |
266 |
32500 |
|
Пиридоксин |
169 |
0,05 М фосфат рН 6,8 |
324 |
7100 |
|
0,1 M NaOH |
308 |
7000 |
Таблица 24
Коэффициенты экстинкции 1 %-ных растворов витаминов для уточнения концентрации стандартных растворов
|
Витамин |
Условия измерения |
l, нм |
Е1 %1 см |
|
Ретинол |
Этанол |
325 |
1832 |
|
a-Токоферол |
Этанол |
292 |
75,8 |
|
b-Каротин |
Этанол |
453 |
2650 |
Таблица 25
Уточнение концентрации стандартных растворов витаминов
|
Витамин |
Молекулярная масса |
Объем стандартного раствора, см3 |
Растворитель |
Объем растворителя, см3 |
|
Аскорбиновая кислота |
176 |
1,0 (рабочий) |
2 %-ная НСl |
2,0 |
|
Тиамин гидрохлорид |
337 |
0,3 |
0,1 М трис-HCl рН 7,5 |
2,7 |
|
Рибофлавин |
376 |
0,1 |
0,1 М фосфат рН 7,0 |
2,9 |
|
Пиридоксингидрохлорид |
206 |
0,1 |
0,05 М фосфат рН 6,8 |
2,9 |
Концентрацию стандартного раствора витамина (мкг/см3) рассчитывают по формуле:
![]() ,
где
,
где
D - поглощение раствора;
Е - коэффициент молярной экстинкции;
М - молекулярная масса;
V - объем раствора в кювете (3 см3), см3;
V1 - объем стандартного раствора, взятого на анализ, см3.
Выделение рибофлавинсвязывающего апобелка из белка куриных яиц
Получение препарата рибофлавинсвязывающего апобелка (РбСБ) проводят при комнатной температуре по компилятивной методике Tillotson J.A., Bashor M.M., White Н.В. с небольшими изменениями.
Реактивы и их приготовление
5 %-ный водный раствор фенола (С6Н5ОН).
5 г фенола растворяют при постоянном перемешивании в дистиллированной воде, доводят объем до 100 см3. Фенол относится к 2 классу опасности.
0,1 н. соляная кислота (НСl).
К дистиллированной воде добавляют 0,8 см3 концентрированной (r = 1,19 г/см3) соляной кислоты по ГОСТ 3118-77, объем доводят до 100 см3.
500 мМ трис-HCl буфер (рН 7,5).
60,5 г трис(гидроксиметил)аминометана (C4H11NO3) растворяют в 500 см3 дистиллированной воды, доводят рН до 7,5 потенциометрически с помощью 1 н. соляной кислоты (примерно 400 см3). Объем буфера доводят дистиллированной водой до 1000 см3.
50 мМ трис-HCl буфер (рН 7,5).
Готовят разведением в 10 раз, добавляя к 100 см3 500 мМ трис-HCl буфер (рН 7,5) дистиллированную воду до 1000 см3.
0,4 М хлорид натрия (NaCl), приготовленный на 50 мМ трис-HCl буфере (рН 7,5).
4,37 г NaCl растворяют в 50 мМ трис-HCl буфере (рН 7,5), доводят объем до 200 см3.
6 мМ соляная кислота (НСl).
К дистиллированной воде добавляют 0,5 см3 концентрированной (r = 1,19 г/см3) соляной кислоты по ГОСТ 3118-77, объем доводят до 900 см3.
250 мМ Na-ацетатный буфер (рН 3,2).
17 г ацетата натрия (CH3COONa) растворяют в 100 см3 дистиллированной воды, доводят потенциометрически рН до 3,2 с помощью ледяной уксусной кислоты (плотность 1,05 г/см3), доводят объем дистиллированной водой до объема 500 см3.
25 мМ Na-ацетатный буфер (рН 3,2).
К 100 см3 250 мМ Na-ацетатного буфера (рН 3,2) добавляют дистиллированную воду, доводят объем до 1000 см3.
25 мМ Na-ацетатный буфер (рН 5,8).
340 мг уксуснокислого натрия (СН3СООNa) растворяют в 40 см3 дистиллированной воды, доводят потенциометрически рН до 5,8 с помощью ледяной уксусной кислоты (плотность 1,05 г/см3). Доливают дистиллированную воду до объема 100 см3.
Выделение
Белки 2 - 10 свежих куриных яиц гомогенизируют вручную в гомогенизаторе Поттера (стекло-тефлон) до получения массы однородной вязкости. Добавляют сухой NaCl до концентрации 20 % (вес/объем) при постоянном перемешивании механической мешалкой. К полученной суспензии приливают равный объем 5 %-ного водного раствора фенола и центрифугируют 10 мин при 1500 g. С помощью ваты снимают верхнюю пленку. Супернатант желтого цвета диализируют 5 ч против 100 объемов дистиллированной воды (5 - 8 смен) и в течение ночи против 20 объемов 50 мМ трис-НСl (рН 7,5), затем центрифугируют.
Надосадочный раствор наносят на колонку (1,2´7 см) DEAE-Sephadex A-50 (Pharmacia, Швеция), предварительно уравновешенную 50 мМ трис-HCl буфером (рН 7,5). После нанесения РбСБ виден в виде желтой зоны в верхней части колонки. Колонку промывают буфером нанесения до тех пор, пока поглощение элюата при 280 нм не становится ниже 0,010. Флавопротеин элюируют 0,4 М NaCl, приготовленным на 50 мМ трис-HCl буфере (рН 7,5). При элюции с колонки собирают визуально видимые ярко окрашенные фракции, содержащие флавопротеин. Фракции флавопротеина в замороженном состоянии при -20 °С могут храниться до 6 мес. Полное отделение рибофлавина от флавопротеина можно проводить двумя способами: диализом против 6 мМ НСl до полного обесцвечивания диализата и хроматографией на колонке КМ-целлюлозы. Для этого элюированные с DEAE-Sephadex A-50 фракции флавопротеина диализируют против 20 объемов 25 мМ Na-ацетатного буфера (рН 3,2) с трехкратной заменой диализирующего буфера в течение 16 ч. Затем наносят на колонку (1´10 см) КМ-целлюлозы (Reanal, Венгрия), предварительно уравновешенную 25 мМ Na-ацетатным буфером (рН 3,2). После нанесения РбСБ также виден на колонке в виде желтой зоны, так как не весь рибофлавин диссоциирован от РбСБ. Для удаления рибофлавина колонку промывают буфером нанесения до полного исчезновения желтой окраски на колонке и уменьшения экстинкции в элюируемом растворе до величины меньше 0,010 при l = 455 нм. Апопротеин элюируют 25 мМ Na-ацетатным буфером (рН 5,8), измеряя поглощение элюата при 280 нм. Фракции с экстинкцией, превышающей 0,1, собирают. Фракции апопротеина, полученные с помощью диализа или хроматографии диализируют против 100 объемов дистиллированной воды в течение ночи, хранят при -20 °С в течение 12 мес. Из белков двух яиц получают 30 - 40 мг апобелка. В лиофилизованном состоянии его активность сохраняется в течение нескольких лет. По данным электрофореза в полиакриламидном геле в присутствии додецилсульфата натрия полученный обоими способами препарат апобелка содержит не менее 80 % пептида с кажущейся молекулярной массой около 36000.
II. Методы определения минеральных веществ
Метод включает подготовку проб и непосредственно проведение количественного определения.
Методы подготовки проб
1.1. Способ сухой минерализации
1.1.1. Способ основан на полном разложении органических веществ путем сжигания пробы в электропечи при контролируемом температурном режиме и предназначен для БАД, кроме содержащих 60 % жира и более.
1.1.2. Подготовка к минерализации
Фарфоровые чашки или тигли после обычной мойки дополнительно обрабатывают раствором уксусной кислоты на кипящей водяной бане в течение 1 ч или промывают горячим раствором азотной кислоты, затем промывают водопроводной водой и ополаскивают дистиллированной водой. Кварцевые чаши и тигли моют раствором азотной кислоты, затем промывают водопроводной и ополаскивают дистиллированной водой.
В чашу (чашку, тигель) в зависимости от определяемого элемента берут навеску продукта из подготовленной к испытаниям пробы, взвешенную с погрешностью 0,01 г при массе навески до 10 г и с погрешностью 0,1 г при массе навески 10 г и более. Масса навески пробы указана в табл. 1 (более подробно см. ГОСТ 26929-94).
1.1.3. Минерализация проб сырья и пищевых продуктов
1.1.3.1. Навеску средней пробы замачивают в 96 % этиловом спирте из расчета 5 см3 спирта на 1 г сухого вещества. Чашки (тигли) с навеской, покрываются часовыми стеклами или чашками Петри, выдерживают при комнатной температуре в течение 12 - 48 ч.
1.1.3.3. БАД с высоким содержанием сахаров перед обугливанием обрабатывают 20 %-ным раствором серной кислоты из расчета 5 см3 кислоты на 1 г сухого вещества. Далее - по п. 1.1.3.2.
1.1.3.4. Чашки с высушенными пробами помещают в холодную электропечь. Минерализацию проб проводят постепенно, повышая температуру электропечи на 50 °С через каждые 30 мин и доводя ее до 450 °С - продолжают минерализацию при этих условиях до получения серой золы.
Чашу с золой вынимают из электропечи, охлаждают до комнатной температуры и серую золу смачивают водой и 0,5 - 1 см3 раствора азотной кислоты (1:1). Затем кислоту досуха выпаривают на электроплитке со слабым нагревом и снова помещают чашу с пробой в электропечь, постепенно доводя температуру до 300 °С, и выдерживают 0,5 ч. Минерализацию считают законченной, когда зола станет белого или слегка окрашенного цвета, без обугленных частиц. При наличии обугленных частиц повторяют обработку золы раствором азотной кислоты или водой, снова доозоляют.
1.1.5. При неполном растворении золы полученной по п. 1.1.4 азотнокислый раствор с осадком упаривают досуха и перерастворяют в минимальном объеме (5 - 10 см3) соляной кислоты (1:1), снова упаривают до влажных солей. Полученные соли растворяют в 1 % соляной кислоте до объема 15 - 20 см3.
1.1.6. Если растворы золы, полученные по п. 1.1.4 или 1.1.5 содержат нерастворимый осадок, объем растворителя можно довести до 25 - 30 см3, пробу подогреть до растворения. Если и в этом случае полного растворения не наблюдается, раствор отфильтровывают через промытый растворителем фильтр и осадок отбрасывают.
1.2. Способ мокрой минерализации
1.2.1. Способ основан на полном разрушении органических веществ пробы при нагревании с серной и азотной концентрированными кислотами с добавлением перекиси водорода и предназначен для всех видов БАД, кроме содержащих сливочное масло и животные жиры.
1.2.2. Подготовка к минерализации
1.2.2.1. Стеклянную посуду после обычной мойки дополнительно промывают раствором азотной кислоты, ополаскивают водопроводной, а затем дистиллированной водой.
1.2.2.2. Навеску жидких и пюреобразных БАД по п. 1.2.2 взвешивают в стакане, переносят в колбу Кьельдаля или плоскодонную колбу, стараясь не попасть на стенки колбы.
1.2.2.3. Навеску БАД по п. 1.2.2 берут на обеззоленный фильтр, заворачивают в него и стеклянной палочкой помещают на дно колбы Кьельдаля или плоскодонной колбы.
1.2.2.4. Навеску сухих БАД (желатиноподобных) помещают в колбу Кьельдаля, добавляют 15 см3 воды, перемешивают. Желатин затем оставляют на 1 ч для набухания.
1.2.3. Минерализация проб сырья и продуктов
1.2.3.1. Кислотная минерализация проб сырья и продуктов, кроме растительного масла, маргарина, пищевых жиров
В колбу с пробой БАД, подготовленной к минерализации по п. 1.2.2, вносят азотную кислоту из расчета 10 см3 на каждые 5 г продукта и выдерживают не менее 15 мин. Затем в колбу вносят 2 - 3 стеклянных шарика для равномерности кипения, закрывают грушевидной пробкой и начинают нагревать на электроплитке слабо, затем сильнее, упаривая содержимое колбы до объема 3 - 5 см3.
Колбу охлаждают, вносят 10 см3 азотной кислоты, содержимое упаривают до объема 5 см3, после чего охлаждают. Эту процедуру повторяют 2 - 4 раза.
В колбу вносят 10 см3 азотной кислоты, 2 см3 серной кислоты, 2 см3 перекиси водорода из расчета на каждые 5 г образца. Минерализацию БАД на молочной основе проводят без добавления серной кислоты. Содержимое колбы упаривают до объема 5 см3, не допуская образования коричневой окраски жидкости. При появлении коричневой окраски нагревание прекращают.
Колбу охлаждают до комнатной температуры, добавляют 5 см3 азотной кислоты и 2 см3 перекиси водорода и снова нагревают до появления белых паров серного ангидрида. Если при этом раствор не обесцветился, эту процедуру повторяют. Минерализацию считают законченной, если раствор после охлаждения остается бесцветным.
Для удаления остатков кислот в охлажденную колбу добавляют 10 см3 воды и кипятят 10 мин с момента выделения белых паров, затем охлаждают. Добавление воды и нагревание повторяют еще 2 раза.
Если при этом образуется осадок, в колбу вносят 20 см3 дистиллированной воды, 2 см3 серной кислоты, 5 см3 соляной кислоты и кипятят до растворения осадка, постоянно дополняя испаряющуюся воду. Полученный минерализат после охлаждения используют для анализа полностью или количественно переносят водой в мерную колбу вместимостью 50 см3, доводят до метки водой и перемешивают.
1.2.3.2. Кислотная минерализация проб с высоким содержанием жира
Исследуемую пробу в колбе Кьельдаля, подготовленную к минерализации по п. 1.2.2, нагревают на электроплитке 7 - 8 ч до образования вязкой массы, охлаждают, добавляют 25 см3 азотной кислоты и вновь осторожно нагревают, избегая бурного вспенивания. После прекращения вспенивания колбу с содержимым охлаждают, добавляют еще 25 см3 азотной кислоты и 12 см3 хлорной кислоты и нагревают до получения бесцветной жидкости. Если во время сжигания жидкость темнеет, то к ней добавляют периодически по 5 см3 азотной кислоты и продолжают нагревание до завершения минерализации. Минерализации. Минерализацию считают законченной, если раствор после охлаждения остается бесцветным. Затем продолжают процедуру по п. 1.2.3.1.
1.3. Способ кислотной экстракции (неполной минерализации)
1.3.1. Способ основан на экстракции элементов из пробы продукта кипячением с разбавленной соляной или азотной кислотой и предназначен для жиросодержащих БАД.
1.3.2. Экстракция и подготовка экстрактов к анализу
1.3.2.1. Экстракция проб БАД
В термостойкую колбу с навеской образца, отобранного по п. 1.2.2, вносят цилиндром 40 см раствора соляной кислоты (1:1) и такой же объем раствора азотной кислоты (1:2).
В колбу добавляют несколько стеклянных шариков, вставляют в нее холодильник, помещают на электроплитку, покрытую асбестом, и кипятят в течение 1,5 ч с момента закипания. Затем содержимое колбы медленно охлаждают до комнатной температуры, не вынимая холодильника.
В колбу с экстракционной смесью жиросодержащих БАД с кислотой помещают в холодную водяную баню для затвердевания жира. Затвердевший жир прокалывают стеклянной палочкой, фильтруют через фильтр, смоченный кислотой, затем промывают фильтр 5 - 7 см3 дистиллированной водой. Оставшийся в колбе жир расплавляют на водяной бане, добавляют 10 см3 раствора кислоты, встряхивают, охлаждают, после охлаждения жир прокалывают и промывную жидкость сливают в тот же сосуд через тот же фильтр, затем фильтр промывают 5 - 7 см3 дистиллированной воды.
Экстракционную смесь масла с кислотой переносят в делительную воронку. Колбу ополаскивают 10 см3 кислоты, которую сливают в ту же воронку. После разделения слоев нижний водный слой сливают через фильтр, смоченный кислотой, в кварцевую или фарфоровую чашку, затем фильтр промывают 5 - 7 см3 дистиллированной воды.
Чашу с экстракционной смесью осторожно выпаривают и обугливают на электроплитке. Затем помещают в электропечь и озоляют до исчезновения обугленных частиц.
1. Атомно-абсорбционный метод определения содержания натрия, калия, кальция, магния, железа, марганца, меди, цинка, свинца, кадмия, кобальта, никеля, хрома
Сущность метода
Метод основан на распылении раствора минерализата испытуемой пробы в воздушно-ацетиленовом пламени. Металлы, находящиеся в растворе минерализата, попадая в пламя, переходят в атомное состояние. Величина адсорбции света с длиной волны соответствующей резонансной линии, пропорциональна значению концентрации металла в испытуемой пробе.
Контрольный (холостой) опыт
При проведении деструкции проб любым способом обязательно проведение контрольного (холостого) опыта для учета уровня загрязнений, которые могут возникнуть на любой стадии подготовки проб. В каждой подготавливаемой серии проб контрольный тигель (чашка, стакан и пр.) обязательно проводят через все стадии подготовки вместе с тиглями, содержащими навески БАД, при одинаковом времени нахождения на электроплитке, в муфеле, при использовании тех же реактивов в том же количестве, той же мерной посуды и т.д. Полученный в холостом опыте раствор анализируют в той же серии измерений, что и растворы проб. Если пробы БАД обычно анализируют в двух повторностях, то контрольный опыт также целесообразно проводить в двух повторностях.
Подготовка к испытанию
Приготовление растворов
Определение содержания элементов в испытуемых растворах проводят методом градуировочного графика, который строится по значениям сигналов абсорбции растворов сравнения.
Головные стандартные растворы (эталоны) готовят из чистых металлов (более 99,9 %) или чистых (х.ч., ос.ч.) устойчивых соединений элементов при использовании растворителей, обеспечивающих устойчивость растворов при хранении (допускается использование коммерческих растворов). Концентрация металла в головном растворе должна быть равна 1 г/дм3 (1000 мкг/см3) (табл. 26). Срок их хранения обычно не превышает 2 - 3 лет, если растворы хранятся в герметичной посуде из стекла твердых сортов, полиэтилена высокой плотности, полистирола, кварца, т.е. материалов, наименее склонных к ионному обмену и абсорбции. Разбавленные растворы с концентрацией порядка 100 мкг/см3 обычно могут храниться до 1 мес, с концентрацией 10 мкг/см3 - 2 - 3 дня. Необходимо избегать применения резиновых пробок, которые могут быть источником загрязнения для ряда микроэлементов, а также избегать хранения растворов в местах, куда попадает прямой солнечный свет. Для приготовления головных стандартных растворов не рекомендуется использовать кристаллогидраты, которые могут не отвечать предполагаемому составу, и нестойкие реактивы, например перманганат калия.
Стандартные растворы сравнения (рабочие эталоны) готовят путем разбавления головных стандартных растворов до концентрации, которая должна соответствовать рабочему диапазону концентраций и быть близка к концентрации элемента в анализируемом растворе. Разбавление производят тем же бланковым раствором, который применяется для приготовления анализируемых растворов проб.
Обычно для построения градуировочного графика в области рабочих концентраций достаточно 3 - 5 растворов сравнения, при работе в линейной области - 2 раствора. При большой и особенно знакопеременной кривизне градуировочного графика необходимо увеличивать число стандартных растворов сравнения или ограничивать область рабочих концентраций более узким диапазоном (табл. 27).
Приготовление растворов и проб к испытанию
Раствор золы может анализироваться непосредственно или после дополнительной подготовки, которая необходима в следующих случаях.
а) Концентрация элемента в растворе значительно превышает верхний предел интервала рабочих концентраций. Производят разбавление исходного раствора бланковым раствором до необходимого уровня концентрации.
б) Концентрация элемента в исходном растворе ниже предела его обнаружения. Если необходима количественная, а не односторонняя («не более»...) оценка, производят концентрирование элемента методом экстракции. Экстракция применяют также при определении микроэлементов на приборах, которые не имеют автоматической коррекции неселективного поглощения.
Для проведения экстракции рассматриваемых элементов могут быть применены разнообразные экстракционные системы, в частности, система диэтилдитиокарбамат натрия (или аммония) - n-бутилацетат (или метилизобутилкетон). Эти системы при регулировании рН позволяют провести экстракцию железа, цинка, меди, марганца, кадмия, свинца, кобальта, никеля и хрома.
Подготовка спектрофотометра к работе и условия измерения
Юстировку прибора проводят в соответствии с технической инструкцией завода (фирмы)-изготовителя. Рекомендуемые условия измерений, а также значения предела определения по данным авторов методики приведены в табл. 27.
Альтернативные длины волн резонансных линий натрия и калия, указанные в табл. 27, могут использоваться для определения в разных областях концентраций, если позволяют технические характеристики лампы и прибора. Для свинца и хрома измерение на указанных альтернативных линиях может отличаться не только чувствительностью, но и отношением сигнал/шум, скоростью дрейфа сигнала. Поэтому выбор резонансной линии в этих случаях проводится для каждого прибора после предварительных сравнительных испытаний.
Указанные в табл. 27 номинальные значения ширины щели монохроматора отвечают отсутствию перекрывания резонансной полосы поглощения с соседними линиями спектра. Тем не менее настройку монохроматора по максимуму излучения источника рекомендуется проводить при минимально возможной щели. Измерения абсорбции таких элементов, как железо, свинец, кадмий, кобальт, никель и хром, при концентрациях, близких к пределу обнаружения, могут проводиться при максимальной щели монохроматора, если для данного прибора это дает улучшение отношения сигнал/шум.
Таблица 26
Рекомендуемые способы приготовления головных стандартных растворов, концентрация металла - 1000 мкг/см3
|
Исходный реактив, навеска, г |
Схема приготовления головного стандартного раствора объемом 1000 см3 |
Концентрация кислоты в конечном растворе |
|
|
1 |
2 |
3 |
4 |
|
Натрий |
NaCl, прокаленный при 500 °С, 2,542 г |
+ Н20 + НСl |
0,02 н. НСl |
|
Калий |
КСl, прокаленный при 500 °С, 1,907 г |
+ Н2О + НСl |
0,02 н. НСl |
|
Кальций |
СаСО3, высушенный при 110 °С, 2,497 г |
+ НСl (2 н), нагрев + Н2О |
|
|
Магний |
Металл, 1,000 г |
+ 100 см3 разбавленной (1:100) НСl, осторожно, по каплям (!) + Н2О |
|
|
MgO, прокаленный при 500 °С, 1,658 г |
+ 25 см3 НСl (25 %) + Н20 |
|
|
|
Железо |
Металл, 1,000 г |
+ 20 см3 НСl (5 н) + 5 см3 концентрированной НNО3 (окисление до Fe3+) + Н2О |
0,1 н. НСl |
|
Цинк |
Металл, 1,000 г |
+ НСl (1:1) + Н20 |
1 н. НСl |
|
ZnO, 1,245 г |
+ HNO3 (40 %) + Н2О |
0,1 н. HNO3 |
|
|
Медь |
Металл, 1,000 г |
+ 50 см3 HNO3 (5 н), упарить досуха на водяной бане + 5 см3 НСl, упарить досуха + разбавленную НСl + Н2О |
0,1 н. НСl |
|
Марганец |
Металл, 1,000 г |
+ 10 см3 HNO3 упаривают досуха на водяной бане + 5 см3 концентрированной НСl, упарить досуха + несколько капель концентрированной НСl + Н2О |
0,02 н. НСl |
|
Свинец |
Металл, 1,000 г |
+ минимальный объем НNО3 (6 н.) + Н2О + НСl |
1 н. НСl |
|
Pb(NO3)2, высушенный при 110 °С, 1,599 г |
+ разбавленную HNO3 (1:100) |
1 % HNO3 |
|
|
Кадмий |
Металл, 1,000 г |
+ разбавленная НNО3, упарить досуха и выдержать при 80 - 90 °С на водяной бане, добавить 5 см3 НСl (1 н.), упарить досуха + разбавленную НСl + Н2O |
1 н. НСl |
|
CdO, 1,142 г |
+ 20 см3 НСl (5 н.) +Н2О + НСl |
1 н. НСl |
|
|
Кобальт |
Металл, 1,000 г |
+ минимальный объем HNO3, упарить досуха, растворить в разбавленной НСl + Н2О |
1 н. НСl |
|
Никель |
Металл, 1,000 г |
+ минимальный объем HNO3, упарить досуха на водяной бане + 5 см3 НСl, упарить досуха + Н2О + НСl |
0,1 н. НСl |
|
Хром |
Металл, 1,000 г, или |
+ НСl + Н2О |
1 н. НСl |
|
К2Сr2О7, высушенный при 150 °С, 2,830 г |
+ разбавленная НСl + Н2О |
0,02 н. НСl |
Таблица 27
Условия атомно-абсорбционных измерений в воздушно-ацетиленовом пламени
|
Длина волны, нм |
Щель, нм |
Пламя |
Высота горелки, мм |
Оптимальный диапазон рабочих концентраций, мкг/см3 |
Предел определения мкг/см3 |
|
|
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
Натрий |
330,2 |
3,0 |
Окислительное |
7 - 8 |
10 - 100 |
1 |
|
589,6 |
0,4 |
7 - 8 |
2 - 30 |
0,1 |
||
|
589,0 |
0,4 |
7 - 8 |
0,5 - 5 |
0,005 |
||
|
Калий |
404,5 |
0,4 |
Стехиометрическое |
7 - 8 |
50 - 1000 |
8 |
|
769,9 |
1,5 |
7 - 8 |
5 - 50 |
0,5 |
||
|
766,5 |
1,5 |
7 - 8 |
0,5 - 10 |
0,005 |
||
|
Магний |
285,2 |
3,0 |
Окислительное |
10 - 15 |
0,1 - 10 |
0,001 |
|
Кальций |
422,7 |
3,0 |
15 - 20 |
5 - 30 |
0,01 |
|
|
Железо |
248,3 |
0,2 |
Стехиометрическое |
7 - 8 |
1 - 10 |
0,01 |
|
Цинк |
213,9 |
1,5 |
Окислительное |
6 - 7 |
1 - 10 |
0,002 |
|
Медь |
324,8 |
1,5 |
Стехиометрическое |
7 - 8 |
0,005 - 5 |
0,003 |
|
Марганец |
279,5 |
0,7 |
7 - 8 |
0,1 - 2 |
0,003 |
|
|
Свинец |
283,3 |
2,0 |
7 - 8 |
0,1 - 2 |
0,02 |
|
|
217,0 |
2,0 |
7 - 8 |
0,1 - 2 |
0,01 |
||
|
Кадмий |
228,8 |
1,0 |
Окислительное |
6 - 7 |
0,02 - 1 |
0,001 |
|
Кобальт |
240,7 |
0,2 |
Стехиометрическое |
6 - 7 |
0,05 - 2 |
0,01 |
|
Никель |
232,1 |
0,2 |
6 - 7 |
0,1 - 5 |
0,01 |
|
|
Хром |
357,9 |
1,3 |
Восстановительное |
6 - 7 |
0,1 - 5 |
0,005 |
|
359,4 |
1,3 |
6 - 7 |
0,05 - 5 |
0,005 |
Три вида воздушно-ацетиленового пламени, указанные в табл. 27, устанавливают приближенно и, как правило, визуально. Восстановительное пламя соответствует соотношению потоков воздух/ацетилен (примерно 4:1), при котором появляется слабое белесое свечение в нижней части факела. Окислительное пламя требует вдвое меньшего расхода ацетилена при том же воздушном потоке. Стехиометрическим пламенем считается промежуточный вариант. При определении микроэлементов, особенно кобальта, никеля и хрома условия измерения могут быть дополнительно улучшены при юстировке соотношения газов и высоты горелки до получения максимального сигнала поглощения (с учетом смещения нулевой линии).
Предел определения соответствовал тройному стандартному отклонению шума измеряемого сигнала прибора у авторов методики.
Особенности определения отдельных элементов
Натрий и калий
Может измеряться как абсорбция, так и эмиссия этих элементов.
В отличие от атомно-абсорбционного измерения эмиссия натрия при длине волны дублетной линии 589 нм может давать завышенные результаты вследствие наложения излучения кальция. Поэтому при атомно-эмиссионных измерениях натрия подготовка проб должна включать отделение кальция посредством осаждения его оксалатом аммония.
При концентрациях калия менее 100 мкг/см3 необходимо согласовывать примерное соотношение калия и натрия в пробах и смешанных растворах сравнения. Это соотношение может устанавливаться как на основе ожидаемых результатов, так и путем предварительных приближенных определений. При концентрациях калия более 100 мкг/см3 влиянием натрия можно пренебречь.
Кальций
Бланковый раствор, растворы проб и растворы сравнения должны содержать 0,5 % стронция (в расчете на металл) в виде хлорида.
Железо и цинк
Растворы проб для анализа во многих случаях требуют разбавления, которое уменьшает межэлементные и матричные влияния. Превышение границ диапазона рабочих концентраций, указанных в табл. 26, может дать смещенные результаты.
Проведение измерений и обработка результатов
Измерения проводят в соответствии с технической инструкцией, прилагаемой к прибору, с учетом следующих особенностей. Потенциальная возможность появления дрейфа или измерения чувствительности вследствие частичного засорения системы распылителя в процессе работы требует более или менее частого контроля, т.е. проведения повторных градуировок. Частота переградуировок определяется скоростью дрейфа и требованиями точности. При отсутствии дрейфа или для кратковременной серии измерений достаточно проведения двух градуировок - в начале и в конце измерений. В других случаях целесообразно проводить измерения по следующей схеме: 1-я градуировка - 1-я серия замеров абсорбции 5 - 10 анализируемых растворов - 2-я градуировка - 2-я серия замеров абсорбции тех же растворов в обратном порядке - 3-я градуировка. Результаты, полученные в двух сериях, усредняют.
Таблица 28
Метрологические характеристики унифицированных атомно-абсорбционных методов анализа БАД
|
X, мг/кг |
r, мг/кг |
R, мг/кг |
Интерполяционные уравнения типа S = аХв |
||||
|
Sr |
sr |
||||||
|
а |
в |
а |
в |
||||
|
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
|
Натрий |
100 |
22 |
95 |
|
|
|
|
|
1000 |
191 |
524 |
0,104 |
0,94 |
1,17 |
0,74 |
|
|
10000 |
1658 |
2848 |
|
|
|
|
|
|
Калий |
100 |
27 |
54 |
|
|
|
|
|
1000 |
148 |
361 |
0,317 |
0,74 |
0,443 |
0,82 |
|
|
10000 |
809 |
2386 |
|
|
|
|
|
|
Кальций |
100 |
29 |
55 |
|
|
|
|
|
1000 |
178 |
512 |
0,267 |
0,79 |
0,223 |
0,97 |
|
|
10000 |
1106 |
4808 |
|
|
|
|
|
|
Магний |
100 |
26 |
53 |
|
|
|
|
|
1000 |
235 |
384 |
0,107 |
0,96 |
0,365 |
0,86 |
|
|
10000 |
2164 |
2755 |
|
|
|
|
|
|
Железо |
10 |
3,8 |
15 |
0,362 |
0,57 |
1,47 |
0,57 |
|
50 |
9,3 |
38 |
|
|
|
|
|
|
100 |
14 |
57 |
|
|
|
|
|
|
200 |
20 |
84 |
|
|
|
|
|
|
Цинк |
1 |
0,34 |
0,73 |
|
|
|
|
|
10 |
2,4 |
4,3 |
|
|
|
|
|
|
50 |
9,6 |
15 |
0,120 |
0,86 |
0,260 |
0,78 |
|
|
100 |
17 |
26 |
|
|
|
|
|
|
Медь |
0,5 |
0,22 |
0,40 |
|
|
|
|
|
1 |
0,31 |
0,64 |
0,106 |
0,41 |
0,228 |
0,67 |
|
|
10 |
0,76 |
3,01 |
|
|
|
|
|
|
30 |
1,2 |
6,3 |
|
|
|
|
|
|
Марганец |
0,1 |
0,056 |
0,12 |
|
|
|
|
|
1 |
0,22 |
0,84 |
0,081 |
0,62 |
0,296 |
0,84 |
|
|
10 |
0,92 |
5,7 |
|
|
|
|
|
|
30 |
1,8 |
14 |
|
|
|
|
|
|
Свинец |
0,01 |
0,005 |
0,014 |
|
|
|
|
|
0,1 |
0,025 |
0,073 |
0,047 |
0,71 |
0,140 |
0,73 |
|
|
0,5 |
0,081 |
0,24 |
|
|
|
|
|
|
1,0 |
0,13 |
0,39 |
|
|
|
|
|
|
Кадмий |
0,01 |
0,0034 |
0,011 |
|
|
|
|
|
0,1 |
0,017 |
0,056 |
0,032 |
0,72 |
0,094 |
0,69 |
|
|
0,5 |
0,055 |
0,17 |
|
|
|
|
|
|
1,0 |
0,090 |
0,27 |
|
|
|
|
|
|
Кобальт |
0,02 |
0,027 |
0,053 |
|
|
|
|
|
0,1 |
0,045 |
0,11 |
0,032 |
0,31 |
0,121 |
0,47 |
|
|
1,0 |
0,090 |
0,34 |
|
|
|
|
|
|
5,0 |
0,15 |
0,72 |
|
|
|
|
|
|
Никель |
0,02 |
0,028 |
0,050 |
|
|
|
|
|
0,1 |
0,084 |
0,14 |
0,134 |
0,66 |
0,210 |
0,63 |
|
|
1 |
0,36 |
0,59 |
|
|
|
|
|
|
10 |
1,7 |
2,49 |
|
|
|
|
|
|
Хром |
0,01 |
0,011 |
0,018 |
|
|
|
|
|
0,1 |
0,045 |
0,11 |
0,070 |
0,63 |
0,243 |
0,77 |
|
|
0,5 |
0,13 |
0,39 |
|
|
|
|
|
|
1,0 |
0,20 |
0,67 |
|
|
|
|
|
Обработка результатов
Пересчет концентрации элемента в растворе (С, мкг/см3) на содержание его в БАД (X, мг/кг) проводят по формуле:
![]() ,
где
,
где
Ск - уровень загрязнения в контрольном опыте, мкг/см3;
K - коэффициент разбавления или концентрирования исходного раствора пробы, равный отношению объема анализировавшегося раствора к объему аликвоты, взятой для разбавления или концентрирования;
Y - объем исходного раствора пробы, см3;
Yк - объем раствора в контрольном опыте, см3;
Р - навеска пробы, г.
Метрологические характеристики
Допустимое расхождение между результатами двух параллельных определений, выполненных в одной лаборатории (r) и е допустимое расхождение между результатами испытаний, выполненных в двух разных лабораториях (R), а также внутрилабораторное среднеквадратичное отклонение (sr) и межлабораторное среднеквадратичное отклонение (SR) приведены в табл. 28, где X - средний уровень концентрации элемента в продукте, мг/кг; а, в - безразмерные эмпирические коэффициенты.
2. Молибдено-ванадиевый метод определения фосфора
Метод предназначен для определения массовой доли фосфора в БАД основан на образовании желтого фосфорно-молибдено-ванадиевого комплекса. Метод заключается в сухой минерализации пробы, растворении золы, проведении цветной реакции с молибдено-ванадиевым реактивом и измерении интенсивности желтого окрашивания раствора.
1. Аппаратура, реактивы и материалы
Калий фосфорнокислый однозамещенный, х.ч., по ГОСТ 4198.
Аммоний ванадиевокислый, мета, х.ч., по ГОСТ 9336.
Аммоний молибденовокислый, ч.д.а., по ГОСТ 3765.
2. Подготовка к испытанию
2.1. Приготовление раствора соляной кислоты с массовой концентрацией 25 г/дм3
В мерную колбу вместимостью 1 дм3 вносят 67,4 см3 концентрированной соляной кислоты и доводят объем раствора до метки дистиллированной водой, перемешивают.
2.2. Приготовление основного стандартного раствора фосфора с массовой концентрацией 1 г/дм3
Раствор готовят по ГОСТ 4212.
Раствор готовят из калия фосфорнокислого однозамещенного, предварительно высушенного до постоянной массы в сушильном шкафу при 104 °С или в эксикаторе над концентрированной серной кислотой. Навеску 4,3930 г КH2РО4 взвешенную с погрешностью не более 0,001 г, растворяют в дистиллированной воде в мерной колбе вместимостью 1 дм3, доводят объем раствора до метки дистиллированной водой и перемешивают.
2.3. Приготовление раствора аммония ванадиевокислого с массовой концентрацией 2,5 г/дм3 (раствор А)
2,5 г аммония ванадиевокислого растворяют в мерной посуде вместимостью 1 дм3 в 500 см3 кипящей дистиллированной воды. После охлаждения раствора до комнатной температуры в него вносят 20 см3 концентрированной азотной кислоты. Объем раствора доводят до 1 дм3 дистиллированной водой, перемешивают.
2.4. Приготовление раствора аммония молибденовокислого с массовой концентрацией 100 г/дм3 (раствор Б)
100 г аммония молибденовокислого растворяют в мерной посуде вместимостью 1 дм3 в 500 см3 дистиллированной воды, подогретой до 50 °С. Раствор охлаждают до комнатной температуры, затем осторожно при перемешивании стеклянной палочкой вносят 100 см3 концентрированной серной кислоты. Перемешивание продолжают до достижения раствором комнатной температуры, доводят его объем до 1 дм3 дистиллированной водой, перемешивают.
2.5. Приготовление молибдено-ванадиевого реактива
Реактив готовят смешиванием равных объемов растворов А и Б. Составной реактив годен в течение месяца.
2.6. Минерализация
Минерализацию проводят сухим способом по ГОСТ 26929-86.
Рекомендуемые величины массы навески указаны в табл. 29.
Таблица 29
Масса навески БАД при определении массовой доли фосфора
|
Масса навески, г |
|
|
БАД на зерновой основе |
3 - 5 |
|
БАД на основе морепродуктов |
5 |
|
БАД на фруктово-овощной основе |
25 |
|
БАД на жировой основе |
1 - 2 |
2.7. Измерения проводят на спектрофотометре в кюветах с расстоянием между рабочими гранями 10 мм при длине волны 436 нм или на фотоэлектроколориметре в кюветах с расстоянием между рабочими гранями 20 мм со светофильтром l = 440,5 нм.
2.8. Приготовление растворов сравнения
2.8.2. При проведении измерений на спектрофотометре в мерные колбы вместимостью 50 см3 вносят 1, 2, 3, 4, 5, 6 см3 рабочего стандартного раствора фосфора, приготовленного по п. 2.8.1, соответственно 100, 200, 300, 400, 500 и 600 мкг фосфора, добавляют воду до объема 10 см3. В каждую колбу вносят 10 см3 разбавленной (1:9) серной кислоты, 10 см3 составного молибдено-ванадиевого реактива, доводят объем раствора до метки дистиллированной водой, перемешивают.
2.8.3. При проведении измерений на фотоэлектроколориметре в кюветах с расстоянием между рабочими гранями 20 мм в мерные колбы вместимостью 50 см3 вносят 0,5; 1; 1,5; 2; 2,5; 3 см3 рабочего стандартного раствора фосфора, приготовленного по п. 3.8.1, соответственно 50, 100, 150, 200, 250 и 300 мкг фосфора, и далее по п. 2.8.2.
2.8.5. Построение аналитического градуировочного графика
Градуировочный график строят, откладывая на оси абсцисс введенные в растворы сравнения массы фосфора в мкг, на оси ординат - соответствующие им значения оптической плотности.
3. Проведение испытания
3.1. Золу, полученную по п. 2.6, растворяют в 5 см3 раствора соляной кислоты (1 + 1) при нагревании на кипящей водяной бане и раствор упаривают до влажных солей, к осадку в тигле добавляют 20 см3 раствора соляной кислоты (25 г/дм3) и нагревают на кипящей водяной бане в течение 5 - 10 мин. После охлаждения содержимое тигля количественно переносят в мерную колбу вместимостью 50 см3 и доводят до метки раствором соляной кислоты 25 г/дм3. Раствор перемешивают.
3.2. В мерную колбу вместимостью 50 см3 помещают аликвотный объем от 1 до 4 см3 раствора, приготовленного по п. 3.1, добавляют воду до объема 10 см3, 10 см3 разбавленной (1:9) серной кислоты, 10 см3 составного молибдено-ванадиевого реактива, доводят объем раствора до метки дистиллированной водой, перемешивают.
Оптическую плотность раствора измеряют по п. 2.8.4.
3.3. По полученному значению оптической плотности с помощью градуировочного графика находят массу фосфора, содержащуюся в аликвотном объеме минерализата.
4. Обработка результатов
4.1. Массовую долю фосфора в мг/100 г вычисляют по формуле:
![]() ,
где
,
где
т - масса фосфора, найденная по градуировочному графику, мкг;
V - общий объем минерализата, см3;
V1 - аликвотный объем минерализата, взятый для испытания, см3;
10 - коэффициент пересчета в мг/100 г;
М - навеска образца, г.
4.2. Результаты определений рассчитывают до третьей значащей цифры.
За окончательный результат испытания принимают среднее арифметическое результатов двух параллельных определений.
4.3. Метрологические характеристики
Относительное допустимое расхождение между результатами двух параллельных определений, выполненных в одной лаборатории, по отношению к среднему арифметическому значению (Rr) и относительное допустимое расхождение между результатами испытаний, выполненных в двух разных лабораториях, по отношению к среднему арифметическому значению (RR), приведены в табл. 30.
Таблица 30
Относительные допустимые внутрилабораторные (Rr) и межлабораторные (RR) расхождения результатов определения
|
Rr, % |
RR, % |
|
|
До 100 |
20 |
40 |
|
От 100 до 300 |
15 |
30 |
|
Свыше 300 |
10 |
20 |
3. Комплексонометрический метод определения кальция и магния
Метод предназначен для определения массовой доли кальция и магния в БАД основан на образовании в щелочной среде малодиссоциированных комплексных соединений катионов кальция и магния с динатриевой солью этилендиаминтетрауксусной кислоты (трилон Б). При рН 12 - 13,5 образуются комплексные соединения кальция, при рН 10,0 - кальция и магния. Метод заключается в сухой минерализации пробы при 450 °С, растворении золы, титровании раствора золы раствором трилона Б в присутствии индикатора кислотного хромового темно-синего.
1. Специфические реактивы
Индикатор хромовый темно-синий кислотный по ТУ 6-09-3870, 5 г/дм3 в растворе этилового спирта (1:5).
Индикатор метилрот по ГОСТ 5853, 1 г/дм3 в растворе этилового спирта (5:3).
Гидроксиламин солянокислый по ГОСТ 5459, х.ч., раствор 100 г/дм3.
Калия или натрия гидроокись по ГОСТ 24363 и ГОСТ 4328, соответственно, х.ч., раствор 200 г/дм3.
Кислота соляная по ГОСТ 3118, х.ч., раствор 25 г/дм3 и разбавленная (1:1).
Натрий сернистый по ГОСТ 2053, ч.д.а., раствор 20 г/дм3.
Натрий лимоннокислый по ГОСТ 22280, х.ч., раствор 100 г/дм3.
Соль динатриевая этилендиаминтетрауксусной кислоты двуводная (трилон Б) по ГОСТ 10652, х.ч., или стандарт-титр.
2. Подготовка к испытанию
2.1. Приготовление растворов для проведения испытания
2.1.1. Приготовление основного раствора кальция с молярной концентрацией 0,01 моль/дм3. Кальций углекислый высушивают до постоянной массы при 100 - 105 °С. Навеску соли 1,009 г, взвешенную с погрешностью не более 0,0002 г, помещают в мерную колбу вместимостью 1000 см3, приливают 20 см3 концентрированной соляной кислоты, доводят объем раствора до 1000 см3 водой, тщательно перемешивают.
2.1.2. Приготовление буферного раствора с рН 10,0
20 г хлористого аммония растворяют в небольшом количестве воды, переносят в мерную колбу вместимостью 1000 см3, приливают 80 см3 аммиака 250 г/дм3, доводят объем раствора до 1000 см3.
2.1.3. Приготовление раствора трилона Б с молярной концентрацией 0,01 моль/дм3
Раствор трилона Б готовят из стандарт-титра. При отсутствии стандарт-титра навеску 3,72 г трилона Б, взвешенную с погрешностью не более 0,01 г, помещают в мерную колбу вместимостью 1000 см3, растворяют в воде, доводят объем раствора до 1000 см3 бидистиллированной водой, перемешивают. Мутный раствор фильтруют. Срок хранения 6 мес.
2.1.4. Определение точной концентрации раствора трилона Б
Пипеткой отбирают 10 см3 основного раствора кальция в коническую колбу вместимостью 100 см3. Вносят 5 - 10 капель индикатора кислотного хромового темно-синего и 6 см3 раствора гидроокиси натрия с массовой концентрацией 200 г/дм3. Быстро титруют из бюретки раствором трилона Б до перехода окраски из малиново-фиолетовой в синюю, не исчезающую в течение 1 мин. Молярную концентрацию раствора трилона Б рассчитывают по формуле:
![]() ,
где
,
где
С - молярная концентрация раствора трилона Б, моль/дм3;
0,01 - молярная концентрация стандартного раствора кальция, моль/дм3;
10 - объем стандартного раствора кальция, взятый для титрования, см3;
V - объем раствора трилона Б, израсходованный на титрование, см3.
2.2. Минерализация
2.2.1. Минерализацию сухим способом проводят по ГОСТ 26929-86.
2.2.2. В табл. 31 указаны рекомендуемые величины массы навесок для некоторых продуктов.
3. Проведение испытаний
3.1. К золе, полученной по п. 2.2, добавляют 20 см3 раствора соляной кислоты с массовой концентрацией 250 г/дм3 и нагревают на кипящей водяной бане в течение 5 - 10 мин до полного растворения золы, количественно переносят в мерную колбу вместимостью 50 см3, доводят объем до метки бидистиллированной водой, перемешивают.
3.2. Перед проведением анализа необходимо приготовить микробюретку для титрования.
3.3. Определение массовой доли кальция
Таблица 31
Масса навески БАД при определении массовой доли кальция и магния
|
Масса навески, г |
|
|
БАД на зерновой основе |
5 - 10 |
|
БАД на основе морепродуктов |
10 - 20 |
|
БАД на фруктово-овощной основе |
25 - 50 |
3.3.2. В конце титрования замечают объем трилона Б, пошедшего на титрование, и прибавляют еще каплю трилона. Если окраска раствора не меняется, титрование считают законченным.
3.3.3. Во время титрования можно ориентироваться на синий ореол, образующийся в месте падения капли трилона. Титрование считают законченным, если синий ореол больше не образуется.
3.3.4. Окраску раствора необходимо смотреть на фоне белого экрана. Окраска оттитрованного раствора устойчива длительное время.
3.3.5. Перед определением кальция оттитровывают контрольную пробу, которую готовят по п. 3.3.1, но вместо раствора золы приливают 4 см3 соляной кислоты с массовой концентрацией 25 г/дм3.
3.3.6. В том случае, если на титрование аликвоты раствора золы уходит больше 3 см3 трилона либо конец титрования затягивается, испытуемый раствор следует разбавить.
3.3.7. Если на титрование уходит меньше 0,5 см3 трилона Б, необходимо увеличить массу навески анализируемой БАД либо аликвотный объем, взятый для анализа. Можно уменьшить концентрацию трилона, разбавив его в 2 раза.
3.4. Определение массовой доли магния
3.4.2. Перед определением магния оттитровывают контрольную пробу, которую готовят по п. 3.4.1, но вместо раствора золы приливают 4 см3 соляной кислоты с массовой концентрацией 25 г/дм3.
3.4.3. Объем трилона Б, пошедший на титрование магния, определяют по разности объемов трилона, пошедшего на титрование, суммы кальция и магния и одного кальция.
Примечание. Метод неприменим для определения массовой доли магния в БАД, в которых массовая доля кальция в 20 раз и более превышает массовую долю магния.
4. Обработка результатов
4.1. Массовую долю кальция (X1) в % вычисляют по формуле:
![]() ,
где
,
где
С - молярная концентрация раствора трилона Б, моль/дм3;
![]() - объем
исходного раствора золы, см3;
- объем
исходного раствора золы, см3;
V1 - объем раствора трилона Б, израсходованный на титрование контрольной пробы, см3;
V2 - объем раствора трилона Б, израсходованный на титрование кальция, см3;
V3 - объем исследуемого раствора, взятый для титрования, см3;
т - навеска БАД, г;
100 - коэффициент пересчета в 100 г образца;
40,08 - атомная масса кальция, г.
4.2. Массовую долю магния (X2) в % вычисляют по формуле:
![]() ,
где
,
где
V4 - объем раствора трилона Б, израсходованный на титрование контрольной пробы, см3;
V5 - объем раствора трилона Б, израсходованный на титрование суммы кальция и магния, см3;
24,305 - атомная масса магния, г;
остальные обозначения те же, что и в формуле (1).
4.3. Вычисления проводят до первого десятичного знака и округляют до целого числа. Минимальная массовая доля кальция, определяемая данным методом в аликвотном объеме минерализата, составляет 10 мкг. Минимальная массовая доля магния, определяемая данным методом в аликвотном объеме минерализата, составляет 6 мкг.
4.4 Метрологические характеристики
Относительно допустимое расхождение между результатами двух параллельных определений, выполненных в одной лаборатории, по отношению к среднеарифметическому значению Rr, и относительно допустимое расхождение между результатами испытаний, выполненных в двух разных лабораториях, по отношению к среднеарифметическому значению RR в зависимости от содержания Са и Mg в продукте приведены в табл. 32.
Таблица 32
Относительно допустимые внутрилабораторные (Rr) и межлабораторные (RR) расхождения результатов определения
|
Содержание Са или Mg, мг на 100 г продукта |
Сходимость, Rr, % |
Воспроизводимость, RR, % |
||
|
Са |
Mg |
Са |
Mg |
|
|
До 10 |
30 |
25 |
60 |
55 |
|
10 - 100 |
20 |
20 |
40 |
40 |
|
Свыше 100 |
15 |
15 |
20 |
20 |
4. Определение свинца и кадмия методом инверсионной вольтамперометрии
Метод основан на использовании инверсионной вольтамперометрии.
Специфические аппаратура, материалы и реактивы
Полярограф марки ПЛС-1 или аналогичный по техническим характеристикам.
Азот газообразный по ГОСТу 9293-74, о.ч., или «0», или другой инертный газ с массовой долей кислорода не более 0,001 %.
Кадмий, ч.д.а.
Дитизон по ГОСТ 10165-79, ч.д.а., растворы в хлороформе 0,01; 0,30 г/дм3.
Кислота азотная, о.ч., по ГОСТ 11135-78 или кислота азотная, х.ч., по ГОСТ 4461-77, плотностью 1,40 г/см3 и разбавленная бидистиллированной водой (1 + 1) и (1 + 2).
Кислота соляная, о.ч., по ГОСТ 14261-77 или кислота соляная по ГОСТ 3118-77, плотностью 1,19 г/см3, разбавленные бидистиллированной водой растворы НСl концентрации 2,0 и 0,1 моль/дм3.
Натрия гидроокись по ГОСТ 4328-77, ч.д.а., гранулированная и раствор NaOH, 0,02 моль/дм3.
Пирогаллол А по ГОСТ 6408-75, ч.д.а., раствор 250 г/дм3.
Ртуть по ГОСТ 4658-73, РО или PI.
Свинец азотнокислый, х.ч., по ГОСТ 4236-77.
Подготовка к испытанию
Подготовка проб
Подготовку проб и минерализацию БАД путем сухого озоления проводят по ГОСТу 26926-86.
Подготовка лабораторной посуды
Лабораторную стеклянную посуду промывают хромовой смесью, водой, азотной кислотой плотностью 1,4 г/см3, несколько раз дистиллированной и дважды бидистиллированной водой и высушивают. Затем промывают раствором дитизона 0,01 г/дм3. Даже при незначительном изменении окраски проводят несколько раз обработку дитизоном: заполняют посуду раствором дитизона 0,30 г/дм3 и выдерживают каждый раз по 30 мин, после чего промывают хлороформом и повторяют обработку, используя раствор дитизона 0,01 г/дм3. Промывают хлороформом и высушивают на воздухе в вытяжном шкафу.
Очистка инертного газа от кислорода
При наличии примеси кислорода более 0,001 % газ пропускают через поглотительную смесь, состоящую из растворов пирогаллола и гидроокиси натрия в соотношении 1:5.
Приготовление основного раствора кадмия
Основной раствор кадмия готовят следующим образом: 1,000 г металлического кадмия помещают в коническую колбу вместимостью 250 см3 и растворяют при нагревании на электроплитке в 25 см3 разбавленной (1:1) азотной кислоты. Раствор выпаривают на электроплитке со слабым нагревом до объема 3 см3, приливают 15 см3 соляной кислоты плотностью 19 г/см3 и вновь выпаривают до того же объема. Выпаривание повторяют еще раза два, добавляя каждый раз по 5 см3 соляной кислоты. После охлаждения добавляют 50 см3 соляной кислоты плотностью 1,19 г/см3, количественно переносят раствор в мерную колбу вместимостью 1000 см3 и доводят бидистиллированной водой до метки. Раствор хранят не более 1 года. Концентрация кадмия в основном растворе равна 1 мг/см3. Стандартные растворы необходимой концентрации готовят последовательным разбавлением в 10, 100 и 1000 раз основного раствора кадмия соляной кислотой 0,1 моль/дм3.
Приготовление основного раствора свинца
Основной раствор свинца готовят следующим образом: свинец азотнокислый перекристаллизовывают и высушивают при 104±1 °С до постоянной массы. 1,599 г высушенной соли растворяют в небольшом объеме бидистиллированной воды и количественно переносят в мерную колбу вместимостью 1000 см3. В колбу добавляют 5 см3 азотной кислоты плотностью 1,4 г/см3 и доводят объем раствора до метки бидистиллированной водой. Раствор хранят не более 1 года. Концентрация свинца в основном растворе равна 1 мг/см3. Стандартные растворы необходимой концентрации готовят последовательным разбавлением в 10, 100 и 1000 раз основного раствора свинца соляной кислотой с концентрацией 0,1 моль/дм3.
Приготовление контрольного раствора
Проверяют каждую новую партию реактивов.
Готовят, используя все реактивы и растворы, аналогично приготовлению испытуемого раствора. Если контрольный раствор содержит измеримое количество кадмия или свинца, его готовят ежедневно при каждой серии измерений.
Подготовка капилляра электрода
В стакан электролизера помещают около 20 см3 раствора соляной кислоты 2 моль/дм3 и проводят сначала накопление в течение 60 с, затем растворение при максимальной скорости развертки (10,5´10 mВ/с). Процедуру электрохимической очистки повторяют еще 2 раза. Ячейку многократно промывают бидистиллированной водой.
Приготовление испытуемого раствора
Золу, полученную по ГОСТ 26929-86, растворяют в тигле при нагревании на электроплитке в 5 см3 разбавленной (1:1) соляной кислоты, раствор выпаривают на электроплитке до объема около 1 см3 и затем досуха на водяной бане. Осадок растворяют в 15 см3 раствора соляной кислоты 0,1 моль/дм3 и количественно переносят в стакан электролизера, смывая тигель 5 см3 той же кислоты.
Проведение испытания
Стакан с пробой помещают в гнездо полярографического датчика, опускают в испытуемый раствор подводящую инертный газ или азот трубку и барбатируют раствор в течение 10 - 15 мин, затем конец поднимают, закрепляют над поверхностью раствора и несколько ослабляют ток газа. Записывают первую полярограмму. При этом измерение проводят инверсионной вольтамперометрией в 3 электродном режиме. Накопление проводят при начальном напряжении 1100 мВ, время накопления от 60 до 300 °С в зависимости от концентрации металлов. Полярограмму записывают при напряжении от минус 1100 мВ до 200 мВ, развертка анодная. Детализацию условий измерений осуществляют в соответствии с инструкцией к прибору и спецификой измеряемой пробы. Затем в стакан с используемым раствором вносят добавку - необходимый стандартный раствор в таком количестве, чтобы высота пика кадмия или свинца примерно удвоилась по сравнению с первоначальной. При этом объем добавляемого стандартного раствора должен быть не более 0,5 см3. Снова пропускают через раствор азот, как указано выше, в течение 5 мин. Затем проводят в тех же режимах запись второй полярограммы.
Обработка результатов
Массовую долю кадмия или свинца (X) в мг/кг или массовую концентрацию (мг/дм3) вычисляют по высоте пиков, измеренных на полярограммах с помощью линейки с точностью до 1 мм.
Расчет проводят по формуле:
для массовой доли:
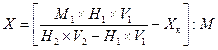 ;
;
для массовой концентрации:
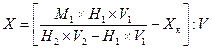 ,
где
,
где
М1 - масса свинца, добавленного перед вторым полярографированием, мкг;
h1 - высота пика свинца, полученного при первом полярографировании, мм;
h2 - высота пика свинца, полученного при втором полярографировании, мм;
V1 - объем испытуемого раствора, приготовленного из озоленной навески, см3;
V2 - объем испытуемого раствора после внесения добавки, см3;
М - масса навески БАД, взятая для озоления, г;
V - объем БАД, взятый для озоления, см3;
Хк - масса свинца в контрольном растворе, мкг.
Вычисление производят до четвертого десятичного знака.
За окончательный результат принимают среднее арифметическое (X) результатов двух параллельных определений.
Метрологические характеристики
Относительное допустимое расхождение между результатами двух параллельных определений, выполненных в одной лаборатории, по отношению к среднему арифметическому значению (Rr) и относительное допустимое расхождение между результатами испытаний, выполненных в двух разных лабораториях, по отношению к среднему арифметическому значению (RR), приведены в табл. 33.
Таблица 33
Относительные допустимые внутрилабораторные (Rr) и межлабораторные (RR) расхождения результатов определения
|
Свинец |
Кадмий |
|
|
Относительная внутрилабораторная сходимость Rr, % |
30 |
30 |
|
Относительное межлабораторное расхождение RR, % |
60 |
|
III. Методы определения микроэлементов
1. Определение йода титрометрическим методом
Принцип метода
Метод основан на взаимодействии йодата калия с йодидом калия в кислой среде (1) и титровании выделившегося йода тиосульфатом натрия (2).
(1) IO3- + 5I- + 6Н + (3I2 + 3Н2О (из соли) (из KI) (из H2SO4)
(2) 2Nа2S2О3 + I2 (2NaI + Na2S4O6 (тиосульфат (иодат (тетратионат натрия) натрия) натрия)
Специфические аппаратура, материалы и реактивы
1 Калий йодистый (KI) по ГОСТ 4232-74.
2 Кислота серная (H2SO4) по ГОСТ 4204-77.
3 Натрий серноватистокислый пятиводный (тиосульфат натрия, Nа2S2О3(5Н2О) по ГОСТ 27068-86 или фиксанал 0,1 г-экв.
4 Крахмал растворимый по ГОСТ 10163-76.
5 Натрия хлорид, ЧДА, ГОСТ 4233-77.
Приготовление реактивов
0,005 М тиосульфат натрия (Na2S2O3(5H2O). 1,24 г Na2S2O3(5H2O) развести в 1000 см3 дистилированной свежепрокипяченной воды. Т.к. кристаллический тиосульфат при хранении набирает влагу, что требует введения поправки на его титр, то в случае возникновения сомнений рекомендуется использовать фиксанал Na2S2O3(5H2O) 0,1 г-эквивалент, который растворяют в дистиллированной воде, доводя конечный объем до 1000 см3 и полученный раствор разводят в 20 раз (50 см3 раствора + 950 см3 воды) до конечной концентрации 0,005 М.
Полученный раствор хранят в прохладном темном месте. Его объем достаточен для анализа 100 - 200 проб в зависимости от содержания в них йода. При соблюдении условий хранения раствор стабилен не менее одного месяца.
2 Н серная кислота (H2SO4). 6 см3 концентрированной H2SO4 медленно доливают в 90 см3 воды, затем доводят раствор водой до конечного объема 100 см3. Полученное количество достаточно для анализа 100 проб. Раствор сохраняет свои свойства неопределенно долгое время.
Примечание. Во всех случаях кислоту надо наливать в воду, а не наоборот, во избежание чрезмерного повышения температуры смеси и разбрызгивания кислоты. Во время добавления кислоты раствор следует непрерывно перемешивать.
10 %-ный йодид калия (KI) свежеприготовленный. 10 г KI растворяют в 100 см3 воды. Хранят в прохладном темном месте. Его количества достаточно для анализа 20 проб.
Насыщенный раствор хлорида натрия (NaCl). В колбу объемом 250 см3 с 80 см3 воды постепенно добавляют при перемешивании и/или нагревании NaCl до тех пор, пока не прекратится его растворение. Хранят под пробкой. Раствор сохраняет свои свойства по крайней мере в течение года.
Индикаторный раствор крахмала. В колбу объемом 250 см3 вносят 1 г растворимого крахмала, добавляют 10 см3 воды и нагревают до растворения крахмала. В полученную горячую смесь добавляют 90 см3 насыщенного раствора NaCl и перемешивают. Полученного объема достаточно для анализа 50 проб. Готовый раствор хранят в прохладном темном месте. Раствор остается стабильным на протяжении месяца.
Примечание. В день проведения анализа раствор необходимо прогреть (но не кипятить) для ресуспендирования возможного осадка.
Проведение анализа
Этап 1. Навеску исследуемой пробы массой 10 г растворяют в 100 см3 дистиллированной воды в конической колбе объемом 250 см3. Если полученный раствор мутный, его необходимо профильтровать.
Этап 2. К полученному раствору добавляют 1 см3 2 н. H2SO4, перемешивают, добавляют 5 см3 10 %-ного раствора KI, перемешивают, закрывают колбу пробкой и помещают на 10 мин. в темное место.
Этап 3. К исследуемому раствору, приобретшему темно-желтую окраску, добавляют из бюретки при перемешивании 0,005 М Na2S2O3 до перехода окраски в светло-желтую. Добавляют в исследуемый раствор примерно 2 см3 индикаторного раствора крахмала, от чего смесь должна приобрести темно-синюю окраску, и продолжают титрование до тех пор, пока последняя не исчезнет.
Отмечают объем раствора тиосульфата, пошедший на титрование.
Требования к безопасности:
· до начала титрования реакционную смесь следует хранить в темном месте ввиду возможности побочного процесса под воздействием света, который вызывает окисление ионов йодида до йода;
· при использовании не вполне остывшего раствора крахмала точность определений понижается;
· если индикаторный раствор добавлен слишком рано, происходит образование прочного, очень медленно реагирующего комплекса йода с крахмалом, что приводит к завышению результатов анализа;
· реакция должна проходить при умеренной комнатной температуре (ниже 30 °С), поскольку йод характеризуется повышенной летучестью, а индикаторный раствор при нагревании теряет чувствительность.
Обработка результатов
Количество йода, мг/кг исследуемой соли вычисляют по формуле:
![]() ,
где
,
где
V - объем 0,005 М Na2S2O3, пошедший на титрование, см3;
10 - навеска соли, взятой на анализ, г;
100 - пересчет на 100 г соли;
0,1057 - количество йода из йодата калия исследуемого образца соли, соответствующее 1 см3 пошедшего на титрование этого образца 0,005 М Na2S2O3.
Примечание. Из уравнений реакций (1) и (2), лежащих в основе данного метода определения йода в соли, обогащенной КIO3, следует, что из общего количества йода, оттитровываемого тиосульфатом натрия в исследуемой пробе, на долю йода, происходящего из КIO3, приходится 1/6 часть, т.е. не 0,6345, а 0,1057 мг.
2. Определение селена спектрофлуориметрическим методом
Принцип метода
Метод основан на флуориметрическом определении комплекса селенистой кислоты с 2,3-диаминонафталином-пиазоселенола, получаемого при мокром сжигании образца смесью азотной и хлорной кислот, с последующим восстановлением соляной кислотой шестивалентного селена до четырехвалентного и образованием комплекса с 2,3-диаминонафталином.
Подготовка к испытанию
Рабочие растворы
0,1 М раствор соляной кислоты: 1 см3 36 % HCL разбавляют до 100 см3 водой.
0,6 М раствор соляной кислоты: 51 см3 36 % HCL разбавляют до 100 см3 водой.
Раствор аммиака: 25 % раствор аммиака разбавляют водой 1:1.
Раствор тетранатриевой соли этилендиамин тетрауксусной кислоты (ЭДТА): 1,25 г ЭДТА растворяют в 100 см3 воды.
Раствор 2,3-диаминонафталина (ДАН) - готовят в день определения: 0,1 г ДАН растворяют в 100 см3 0,1 М соляной кислоты при слабом нагревании. Раствор трижды экстрагируют гексаном, фильтруют через складчатый фильтр и хранят под слоем гексана в темноте при температуре 0 - 4 °С до момента флуориметрического определения. Не допускается облучение прямым светом и использование смазки для шлифов.
Основной стандартный раствор, содержащий 1 мкМ селена/см3:
0,01729 г Nа2SеО3 растворяют в небольшом количестве 0,1 М соляной кислоты в мерной колбе на 100 см3, доводят объем до метки соляной кислотой этой же концентрации. Раствор хранят в темноте не более 1 месяца.
Рабочий стандартный раствор, содержащий 1 нМ селена/см3: пипеткой вносят 5 см3 основного стандартного раствора в мерную колбу на 500 см3 и доводят объем до метки 0,1 М соляной кислотой. Раствор хранят в темноте не более 1 месяца.
Мокрое сжигание. Взвешивают и переносят в пробирки для сжигания от 50 до 100 мг БАД и соответствующий образец сравнения. Для построения калибровочного графика в 7 пробирок вносят: 0 (контроль), 0,1; 0,2; 0,4; 0,5; 0,6 мл стандартного рабочего раствора, что соответствует 0; 0,1; 0,2; 0,4; 0,5; 0,6 нМ селена в пробе. Во все пробирки добавляют 1,5 мл смеси, содержащей азотную и хлорную (42 %-ные) кислоты в соотношении 1:0,7 и оставляют при комнатной температуре на 12 - 18 час., затем пробирки помещают в блок для сжигания и минерализуют при 120 °С - 1 час., 150 °С - 1 час., 180 - 185 °С - 1,5 часа. При наличии обугливания сжигание повторяют с новой навеской, увеличив количество хлорной кислоты до 1,0 - 1,4 см3.
Удаление следов азотной кислоты
Блок сжигания охлаждают до 150 °С, добавляют к пробам 1 - 2 капли перекиси водорода и выдерживают при указанной температуре в течение 10 мин. Затем пробы сразу же вынимают из блока сжигания.
Восстановление селената
К пробам добавляют по 1 см3 6 М соляной кислоты и выдерживают при 110 °С в течение 10 мин. Затем сразу же вынимают из блока сжигания и добавляют 1 см3 дистиллированной воды.
Конденсация селенистой кислоты с ДАН
В каждую пробирку добавляют по 0,5 см3 раствора ЭДТА и по 1 см3 раствора аммиака, быстро доводят рН до 1 - 2 по универсальному индикатору, добавляя при необходимости по каплям раствор аммиака или 0,1 М соляную кислоту. К полученным растворам приливают по 1 см3 раствора ДАН и выдерживают 30 мин при 50 - 55 °С, закрыв предварительно пробирки от прямого света. По окончании реакции пробы охлаждают до комн. температуры и каждую интенсивно встряхивают с 2,5 см3 гексана в течение 50 - 60 сек.
Спектрофлуориметрический анализ
Определение интенсивности флуоресценции осуществляют при длине волны возбуждающего света 376 нм и эмиссионного света 519 нм. Дополнительный контроль за полнотой сжигания осуществляют по спектрам флуоресценции в диапазоне 450 - 600 нм. Полученный спектр должен иметь один максимум 519 нм.
Обработка результатов
Содержание селена, мкг/кг или мкг/л (X) вычисляют по формуле:
Х = 79 ´ С/а, где
С - количество селена, нМ, в пробе, определенное по калибровочному графику;
79 - атомная масса селена;
а - навеска образца, г или см3.
За окончательный результат принимается среднее арифметическое двух параллельных определений.
Метрологические характеристики
Относительное стандартное отклонение в интервале 1 - 600 мкг/кг - не более 10 %.
3. Определение токсичных элементов
Анализируют согласно методов главы 2, раздела II и согласно нормативным документам «Сырье и продукты пищевые. Подготовка проб. Минерализация для определения содержания токсичных элементов», межгосударственный стандарт ГОСТ 26929-94, ГОСТ 26927-86 «Сырье и продукты пищевые. Атомно-абсорбционный метод определения токсичных элементов», межгосударственный стандарт ГОСТ 30178-96 «Методические указания по обнаружению и определению общей ртути в пищевых продуктах методом беспламенной атомной абсорбции № 5178-90», ГОСТ 26930-86 «Сырье и продукты пищевые. Метод определения мышьяка».
Глава 3
МИНОРНЫЕ БИОЛОГИЧЕСКИ АКТИВНЫЕ КОМПОНЕНТЫ БАД
(методы определения подлинности БАД)
1. Определение антоцианинов
Антоцианины являются водо-растворимыми пигментами, обусловливающими красную, синюю и фиолетовую окраску фруктов. Они относятся к классу флавоноидов и представляют собой гликозиды катиона флавония (рис. 1).
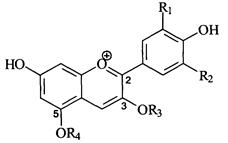
Рис. 1.
|
r1 |
R2 |
Агликон |
Сокращенное название |
|
н |
Н |
пеларгонидин |
Pgd |
|
он |
н |
цианидин |
Cyd |
|
ОМе |
н |
пеонидин |
Pnd |
|
ОН |
он |
дельфинидин |
Dpd |
|
ОМе |
он |
петунидин |
Ptd |
|
ОМе |
ОМе |
мальвидин |
Mvd |
|
R3 и R4 - Н или гликозид |
|||
1.1. Методика определения качественного и количественного состава антоцианиновых пигментов с помощью ВЭЖХ
Образцы БАД с антоцианин-содержащими компонентами (экстракты черники, красного винограда и т.д.) экстрагируют дистиллированной водой; образцы соковых концентратов или сиропов разбавляют дистиллированной водой таким образом, чтобы суммарная концентрация антоцианинов составляла 0,05 - 0,2 мг в см3. Пробы фильтруют через мембранный фильтр Zetapor с диаметром пор 0,2 мкм. Разделение проводят на хроматографической колонке PLRP-S (полистирол-дивинилбензол), PolymerLabs, Великобритания), 100 А, 5 мкм, 250´4,6 ID мм, элюент: 4 об. % ортофосфорная кислота, ацетонитрил, 90:10 об. %, скорость подачи элюента 1,0 см3/мин. Детектирование фотометрическое, l = 520 нм.
Идентификацию пиков проводят путем сравнения с хроматограммами ягод с известным составом антоцианиновых пигментов и с литературными данными. Относительное содержание индивидуальных пигментов определяют как отношение площади хроматографического пика и суммы площадей пиков всех идентифицированных антоцианинов. Факторы удерживания антоцианинов приведены в табл. 34.
Таблица 34
Факторы удерживания антоцианиновых пигментов
|
Антоцианины |
k |
||
|
1 |
Cyd-3-sop-5-glu |
цианидин -3-софорозид-5-глюкозид |
1,9 |
|
2 |
Dpd-3-gal |
дельфинидин-3-галактозид |
3,0 |
|
3 |
Dpd-3-glu |
дельфинидин-3-глюкозид |
3,6 |
|
4 |
Cyd-3-sop |
цианидин-3-софорозид |
3,9 |
|
5 |
Cyd-3-glu-rut |
цианидин-3-глюкорутинозид |
4,0 |
|
6 |
Dpd-3-rut |
дельфинидин-3-рутинозид |
4,2 |
|
7 |
Cyd-3-gal |
цианидин-3-галактозид |
5,2 |
|
8 |
Dpd-3-ara |
дельфинидин-3-арабинозид |
5,5 |
|
9 |
Cyd-3-glu |
цианидин-3-глюкозид |
5,9 |
|
10 |
Cyd-3-xyl-rut |
цианидин-3-ксилозорутинозид |
6,1 |
|
11 |
Cyd-3-rut |
цианидин-3-рутинозид |
7,0 |
|
12 |
Ptd-3-gal |
петунидин-3-галактозид |
7,5 |
|
13 |
Cyd-3-ara |
цианидин-3-арабинозид |
7,9 |
|
14 |
Ptd-3-glu |
петунидин-3-глюкозид |
8,4 |
|
15 |
Pgd-3-glu |
пеларгонидин-3-глюкозид |
9,6 |
|
16 |
Pnd-3-gal |
пеонидин-3-галактозид |
10,0 |
|
17 |
Pgd-3-ara |
пеларгонидин-3-арабинозид |
11,9 |
|
18 |
Pnd-3-glu |
пеонидин-3-глюкозид |
12,3 |
|
19 |
Mvd-3-glu |
мальвидин-3-глюкозид |
16,1 |
|
20 |
Pnd-3-ara |
пеонидин-3-арабинозид |
16,2 |
На рис. 2 приведена хроматограмма антоцианинов сока черники, для которой характерен самый богатый профиль пигментов.
Систематизированные литературные и экспериментальные данные по составу антоцианиновых пигментов окрашенных соков представлены в табл. 35, являются основой при оценке подлинности и качества соков, сокосодержащей продукции и БАД.
1.2. Суммарное содержание антоцианиновых пигментов
Суммарное содержание антоцианиновых пигментов определяли методом рН-дифференциальной спектрофотометрии. Известно, что поглощение антоцианиновых пигментов сильно зависит от рН раствора: в сильнокислой среде окраска большинства антоцианинов ярко-красная, при увеличении рН она постепенно переходит в темно-синюю. Это связано с изменением структуры входящего в состав пигмента агликона. Различие в адсорбции при l = 510 нм и рН 1 и 4,5 пропорционально содержанию антоцианина. Так как различие в величинах молярной адсорбции индивидуальных антоцианинов незначительно, суммарную концентрацию пигментов можно определять относительно одного из них, например, цианидин-3-глюкозида (e = 26900):
![]() ,
,
![]() , где
, где
А - поглощение,
e и MW - коэффициент молярного поглощения и молекулярная масса антоцианина, используемого в качестве стандарта (для цианидин-3-глюкозида 26900 и 449,2 соответственно),
AF - разведение,
AV - объем, мл,
Al - длина кюветы, см,
т - масса образца, мг.
Метрологическая характеристика метода
Предел обнаружения метода 0,001 %. Стандартное отклонение сходимости (Sr) не превышает 0,1 - 0,15. Среднее значение открываемости - 92 - 95 %.
Рис. 2. Хроматограмма антоцианинов черничного сока. Номера пиков соответствуют номерам антоцианинов в табл. 34.
Систематизированные литературные и экспериментальные данные по составу антоцианиновых пигментов окрашенных соков представлены в табл. 35, являются основой при оценке подлинности и качества соков, сокосодержащей продукции и БАД.
Таблица 35
|
Относительное содержание антоцианинов, % |
||||||||||||||||||||
|
Cyd-3-sop-5-glu |
Dpd-3-gal |
Dpd-3-glu |
Cyd-3-sop |
Cyd-3-glu-rut |
Dpd-3-rut |
Cyd-3-gal |
Dpd-3-ara |
Cyd-3-glu |
Cyd-3-xyl-rut |
Cyd-3-rut |
Ptd-3-gal |
Cyd-3-ага |
Ptd-3-glu |
Pgd-3-glu |
Pnd-3-gal |
Pgd-3-ага |
Pnd-3-glu |
Mvd-3-glu |
Pnd-3-аrа |
|
|
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
10 |
11 |
12 |
13 |
14 |
15 |
16 |
17 |
18 |
19 |
20 |
21 |
|
Гранат |
30 |
24 |
|
21 |
|
|
|
|
25 |
|
|
|
|
|
|
|
|
|
|
|
|
Черника |
|
10 |
14 |
|
|
|
8 |
9 |
18 |
|
|
10 |
9 |
3 |
|
|
|
8 |
10 |
|
|
Красный виноград |
|
|
22 |
|
|
|
|
|
3 - 2 |
|
|
|
|
20 - 22 |
|
|
|
4 - 5 |
52 - 38 |
|
|
Черная смородина |
|
|
18 |
|
|
44 |
|
|
6 |
|
32 |
|
|
|
|
|
|
|
|
|
|
Красная смородина |
|
|
|
1 |
12 |
|
|
|
1 |
70 |
10 |
|
|
|
|
|
|
|
|
|
|
Слива |
|
|
|
|
|
|
|
|
53 |
|
47 |
|
|
|
|
|
|
|
|
|
|
Клюква |
|
|
|
|
|
|
16 |
|
4 |
|
|
|
20 |
|
|
33 |
|
10 |
|
17 |
|
Клубника |
|
|
|
|
|
|
|
|
2 |
|
|
|
|
|
92 |
|
6 |
|
|
|
|
Черешня |
|
|
|
15 |
|
|
|
|
45 |
|
40 |
|
|
|
|
|
|
|
|
|
|
Вишня |
|
|
|
2 |
74 |
|
|
|
4 |
|
20 |
|
|
|
|
|
|
|
|
|
|
Малина |
|
|
|
25 |
6 |
|
|
|
39 |
|
6 |
|
|
|
|
|
|
|
|
|
|
Ежевика |
|
|
|
|
|
|
|
|
94 |
|
|
|
|
|
|
|
|
1 |
|
|
|
Брусника |
|
|
|
|
|
|
83 |
|
5 |
|
|
|
12 |
|
|
|
|
|
|
|
|
Арония |
|
|
|
|
|
|
60 |
|
2 |
|
|
|
32 |
|
|
|
|
3 |
|
|
|
Крыжовник черный |
|
|
|
|
|
4 |
|
|
14 |
|
80 |
|
|
|
|
|
|
|
|
|
|
Калина |
|
|
|
|
|
|
98 |
2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
Ирга |
|
|
|
|
|
|
75 |
|
17 |
|
|
|
|
8 |
|
|
|
|
|
|
2. Определение органических кислот с помощью ВЭЖХ
Принцип метода
Органические кислоты определяют в условиях обращеннофазной высокоэффективной жидкостной хроматографии (ОФ ВЭЖХ). Разделение органических кислот происходит в хроматографической колонке, наполненной октадецилсиликагелем. Концентрации определяют с помощью спектрофотометрического детектора при l = 210 нм по методу внешнего стандарта.
Приборы и реагенты
Хроматографическая система: насос высокого давления Altex 110А (США), инжектор Rheodyne 7125 (США), колонка Kromasil С18, 5 мкм, 250´4,6 ID мм (MetaChem, США), спектрофотометрический детектор Spectroflow 757 (Kratos, США), система для обработки хроматографических данных МультиХром 1,5х (Амперсенд, Россия).
Подвижная фаза: 0,2 М фосфатный буфер, рН 2,6. Объемная скорость: 1,0 см3/мин. Перед использованием, подвижную фазу фильтруют через нейлоновый фильтр (диаметр пор 0,45 mм) или бумажный фильтр («синяя лента»).
Реагенты: калия дигидрофосфат (КH2РО4), фосфорная кислота (Н3РO4, 85 %), ацетонитрил (CH3CN), дистиллированная вода.
Приготовление проб и стандартов
Стандартные растворы кислот готовят путем растворения точных навесок соответствующих кислот (purum, ³ 99 %, Fluka или Sigma) в мерных колбах. Градуировочные графики строят в координатах S (площадь пика) - С (концентрация кислоты), г/дм3 в интервале концентраций 0,1 - 30 г/дм3 для яблочной кислоты, 0,1 - 40 г/дм3 для лимонной кислоты, 0,1 - 1 г/ дм3 для аскорбиновой кислоты, 0,05 - 0,5 г/дм3 для изолимонной кислоты, 0,01 - 0,1 г/дм3 для фумаровой кислоты. Растворы фильтруют через нейлоновый фильтр (диаметр пор 0,45 мкм) или бумажный фильтр («синяя лента»).
Пробы готовят растворением сухих или разбавлением жидких концентратов БАД в 10 - 25 раз дистиллированной водой. Приготовленные растворы фильтруют через нейлоновый фильтр (диаметр пор 0,45 мкм) или бумажный фильтр («синяя лента»).
ВЭЖХ определение органических кислот
Перед проведением анализа хроматографическую колонку кондиционируют подвижной фазой ацетонитрил: вода (70:30 об. %), затем систему промывают фосфатным буфером до установления ровной базовой линии. Стандартные растворы (5 - 20 мкл) вводят в хроматограф попеременно через каждые 2 - 3 пробы (10 мкл). Порядок элюирования органических кислот в условиях ОФ ВЭЖХ следующий:
Винная < хинная < янтарная < гидроксилимонная < яблочная < изолимонная < шикимовая < аскорбиновая < фумаровая < лимонная
Коэффициенты емкости основных органических кислот, определяемых в БАД на основе фруктов и ягод, представлены в табл. 36.
Таблица 36
|
k¢ |
|
|
Винная |
0,4 |
|
Хинная |
0,6 |
|
Янтарная |
0,8 |
|
Гидроксилимонная |
1,1 |
|
Яблочная |
1,2 |
|
Изолимонная |
1,4 |
|
Шикимовая |
1,5 |
|
Аскорбиновая |
1,8 |
|
Фумаровая |
3,9 |
|
Лимонная |
4,4 |
Концентрации органических кислот в пробах рассчитываю по площадям или высотам хроматографических пиков.
Примеры хроматограмм органических кислот апельсина и клюквы приведены на рис. 3 и 4.
Рис. 3. Органические кислоты апельсина.
Рис. 4. Органические кислоты клюквы.
3. Определение 5-оксиметилфурфурола в БАД на основе меда и углеводных сиропов
Принцип метода
Метод определения массовой концентрации 5-оксиметилфурфурола в соках, напитках и меде основан на применении высокоэффективной жидкостной хроматографии (ВЭЖХ).
Метод включает следующие этапы:
· разбавление аликвоты или навески жидкого БАД дистиллированной водой; растворение навески меда или медосодержащего БАД в дистиллированной воде;
· фильтрование полученного раствора;
· обнаружение и количественное определение ОМФ с помощью ВЭЖХ с УФ-детектором с использованием метода внешнего стандарта.
Средства измерений, вспомогательное оборудование, материалы и реактивы
Жидкостной хроматограф с насосом высокого давления с подачей растворителя от 0,1 до 5,0 см3/мин., оборудованный спектрофотометрическим детектором с переменной длиной волны и системой для сбора и обработки хроматографических данных Мультихром, версия 1,5х (Амперсенд, Россия).
Колонка и предколонка хроматографические с силикагелем, химически связанным с октадецилсиланом (силикагель С18) с размером частиц 5 мкм; длина колонки - 25 см, предколонки - 4,5 см, внутренний диаметр колонок - 0,46 см;
Микрошприцы МШ-10 и МШ-25 для жидкостной хроматографии.
Спектрофотометр СФ-26, сф-46 или подобный, позволяющий проводить измерения при длинах волн 200 - 350 нм, с допустимой абсолютной погрешностью измерений коэффициента пропускания не более 1 %.
Аппарат для встряхивания проб типа АВУ-6С, ТУ 64-1-2451-78.
Весы лабораторные общего назначения по ГОСТ 24104 с наибольшим пределом взвешивания 200 г и погрешностью ±0,0001 г.
Весы лабораторные с наибольшим пределом взвешивания до 200 г, с пределом допустимой погрешности ±0,0005 г по ГОСТ 2404-80.
Дистиллятор.
Колбы мерные наливные 2-50-2, 2-100-2, 2-250-2, 1-1000-2 по ГОСТ 1770.
Пипетки 4-1-2 или 5-1-2, 4-2-10 или 5-2-10, 4-2-25 или 5-2-25 по ГОСТ 29227.
Термометр жидкостный стеклянный с пределами измерения 0 - 100 °С и ценой деления 1 °С по ГОСТ 28498.
Ацетонитрил, ч., ТУ 6-09-3534-74.
Фильтры обеззольные ФО-ФС-15 «Синяя лента» по ТУ 2642-001-42624157-98;
Вода дистиллированная, ГОСТ 6709.
Калий железистосинеродистый 3-водный по ГОСТ 4207, х.ч., раствор концентрацией 150 г/дм3 (раствор Карреза 1);
Цинк уксуснокислый 2-водный по ГОСТ 5823, ч.д.а., раствор концентрацией 300 г/дм3 (раствор Карреза II);
Фильтр нейлоновый имп. на шприце в обойме 25 мм, размер пор 0,45 мкм;
Бензол для хроматографии, х.ч. по ТУ 6-09-779-76;
Метанол - яд для жидкостной хроматографии, ос.ч. по ТУ 6-09-14-2192-85;
Кислота уксусная > 99,8 %, ос.ч., ГОСТ 18270-72;
Баллон с сжатым азотом, ос.ч. по ГОСТ 9293-74;
5-оксиметилфурфурол (ОМФ) кристаллический, перекристаллизованный из метанола при температуре 64 - 65 °С и атмосферном давлении.
Подготовка к проведению измерений
Приготовление стандартного раствора 5-оксиметилфурфурола
Приготовление основного стандартного раствора 5-ОМФ
0,05 г кристаллического 5-оксиметилфурфурола количественно переносят в мерную колбу вместимостью 250 см3, растворяют в 25 см3 ацетонитрила, доводят бензолом до метки и тщательно перемешивают. Получают стандартный раствор в смеси бензол-ацетонитрил (9:1) с массовой долей 5-оксиметилфурфурола 0,2 мкг/см3.
Приготовление рабочего стандартного раствора 5-ОМФ
Аликвоту в 1 см3 основного стандартного раствора ОМФ переносят в мерную колбу вместимостью 100 см3, растворитель отдувают досуха в токе азота, доводям метанолом до метки и тщательно перемешивают. Получают рабочий стандартный раствор ОМФ концентрацией 0,002 мкг/см3, который используют при градуировке хроматографа.
Основной и рабочий стандартные растворы 5-оксиметилфурфурола хранят в стеклянной посуде (пикнометре, мерной колбе) с притертой пробкой в прохладном месте (при температуре около 0 °С). Сроки годности основного раствора 1 год, рабочего - 1 неделя.
Установление массовой доли 5-оксиметилфурфурола в рабочем стандартном растворе
Установление точной массовой доли 5-оксиметилфурфурола в рабочем стандартном растворе производят с помощью УФ-спектрофотометрии согласно данным о коэффициенте молярной экстинкции 5-оксиметилфурфурола в максимуме поглощения в УФ-спектре. Снимают УФ спектр рабочего стандартного раствора в метаноле и измеряют его оптическую плотность. Рассчитывают концентрацию ОМФ в рабочем стандартном растворе по формуле:
![]() ,
где
,
где
С - концентрация исследуемого раствора, мкг/см3 или нг/мкл,
D - оптическая плотность раствора;
М - молекулярная масса 5-оксиметилфурфурола (126);
Е - коэффициент молярной экстинкции для метанольного раствора (16830);
L - толщина слоя раствора в см.
Значение относительной погрешности при определении массовой доли 5-оксиметилфурфурола в растворе с помощью УФ-спектрофотометрии не превышает 5 % для доверительной вероятности 0,95.
Приготовление подвижной фазы
В мерную колбу вместимостью 1000 см3 помещают 150 см3 метанола, примерно 500 см3 бидистиллированной воды и 10 см3 уксусной кислоты, перемешивают и выдерживают до комнатной температуры, затем доводят до метки бидистиллированной водой, тщательно перемешивают.
Подготовка образца
10 см3 сокосодержащего БАД или концентрата фруктового сока помещают в мерные колбы вместимостью 100 см3 или 250 см3, доводят дистиллированной водой до метки и тщательно перемешивают. Полученные растворы фильтруют через бумажный фильтр «синяя лента» или нейлоновый фильтр.
5 - 10 г меда, медосодержащего пищевого продукта или БАД помещают в мерные колбы вместимостью 100 см3 или 250 см3, добавляют около 50 см3 дистиллированной воды, перемешивают до растворения, последовательно добавляют по 5 см3 растворов Карреза I и Карреза II, каждый раз перемешивая смесь, доводят дистиллированной водой до метки и тщательно перемешивают. Фильтруют через бумажный фильтр «синяя лента» или нейлоновый фильтр. Аликвоту полученных фильтратов вводят в хроматограф.
Выполнение измерений и вычисление результатов анализа
Условия хроматографического анализа
Подвижная фаза метанол-вода-уксусная кислота (15:84:1), скорость потока 1,0 мл/мин., обнаружение по поглощению в УФ-области спектра при длине волны 284 нм, чувствительность устанавливают 0,01 единиц адсорбции на всю шкалу, вводимый объем образца 5 - 15 мкл.
Вычисление результатов анализа
Массовую концентрацию 5-оксиметилфурфурола рассчитывают по формуле:
![]() ,
где
,
где
М - массовая концентрация 5-оксиметилфурфурола, мг/дм3 или мг/кг (ррm),
m - массовая доля внешнего стандарта ОМФ, введенного для калибровки, хроматографа, нг,
Нобр. - высота пика исследуемого компонента, соответствующая определенной массовой концентрации компонента в исследуемом образце, мм;
Нст - средняя высота пика внешнего стандарта ОМФ, введенного для калибровки хроматографа, мм;
![]() - общий объем анализируемой пробы, мкл;
- общий объем анализируемой пробы, мкл;
![]() - объем аликвоты, введенной в хроматограф, мкл;
- объем аликвоты, введенной в хроматограф, мкл;
Р - навеска или объем пробы, см3 или г.
Вычисления проводят до второго десятичного знака. За результат принимают среднее арифметическое результатов двух параллельных измерений и выражают целым числом с одним десятичным знаком.
Основные характеристики метода
Предел обнаружения метода составляет 1,0 мг/кг.
Диапазон определяемых концентраций от 1,0 до 1000 мг/кг.
При соблюдении всех регламентируемых методикой условий проведения измерений относительная характеристика погрешности (стандартное отклонение сходимости) результата анализа при концентрации ОМФ в жидких БАД 2,0 мг/л не превышает 0,124 (3,43 %) и при концентрации ОМФ в меде и медсодержащих БАД 5,0 мг/кг - 0,072 (5,34 %).
Среднее значение открываемости ОМФ при концентрациях от 2,0 до 25 мг/кг составляет 87 - 95 %.
4. Определение состава моно- и дисахаридов с помощью ВЭЖХ
Принципиальная схема метода
Метод заключается в пробоподготовке путем разбавления дистиллированной водой образцов жидких БАД или экстракции сухих БАД дистиллированной водой до концентрации сахаров примерно 2 - 10 мг/мл, фильтрования полученного раствора, разделении и количественном определении сахаров с помощью ВЖХ.
Приготовление стандартных растворов
Аналитические образцы фруктозы, глюкозы, сорбита и сахарозы сушат до постоянного веса согласно ГОСТ 12570-67 «Сахар-песок и сахар-рафинад. Метод определения содержания влаги».
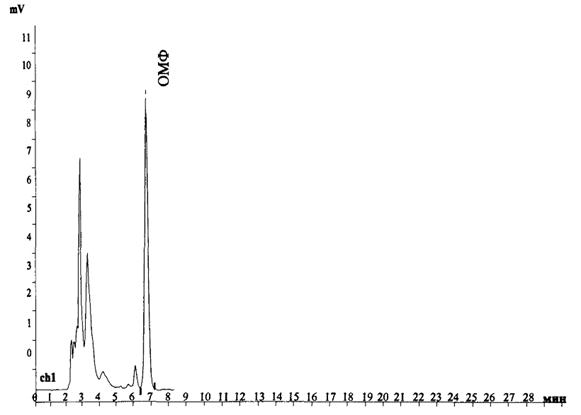
Рис. 5. Сироп с 5 мл/100 мл С (5 - ОМФ) = 9,2 мг/л
Готовят стандартные растворы фруктозы, глюкозы, сорбита и сахарозы с концентрациями от 3 до 10 мг/см3 в зависимости от чувствительности рефрактометрического детектора. Для этого навески сахаров массой 1±0,001 г или 0,3±0,001 г помещают в мерные колбы вместимостью 100 см3, доводят бидистиллированной или дистиллированной фильтрованной через капроновый фильтр марки 0,45 мкм RC водой до метки и тщательно перемешивают.
Готовят стандартный раствор смеси фруктозы, глюкозы и сахарозы, с массовыми долями каждого компонента 10 мг/см3 или 3 мг/см3. Для этого навески сахаров массой 1±0,001 г или 0,3±0,001 г помещают в одну мерную колбу вместимостью 100 см3, доводят бидистиллированной или дистиллированной фильтрованной через капроновый фильтр марки 0,45 мкм RC водой до метки и тщательно перемешивают.
Приготовление исследуемого образца
Анализируемый образец БАД растирают в ступке. Навеску массой 5±0,01 г помещают в мерную колбу вместимостью 250 см3, добавляют 150 см3 подогретой до 75 - 80 °С дистиллированной воды и встряхивают на аппарате для встряхивания 20 - 30 мин. Добавляют 15 см3 30 %-ного раствора ацетата цинка, встряхивают, охлаждают до комнатной температуры, доводят дистиллированной водой до метки и тщательно перемешивают. Фильтруют через капроновый фильтр марки 0,45 мкм RC или складчатый бумажный фильтр «синяя лента». Аликвоту фильтрата вводят в хроматограф.
20 см3 сокосодержащего БАД или 10 г углеводсодержащего сиропа помещают в мерную колбу вместимостью 100 см3, доводят бидистиллированной водой до метки, тщательно перемешивают и фильтруют через капроновый фильтр марки 0,45 мкм RC или через складчатый бумажный фильтр обеззоленный марки ФОМ. Аликвоту фильтрата вводят в хроматограф.
Анализ с помощью ВЭЖХ
Анализ ВЭЖХ с использованием амино фазы
Условия ВЭЖХ: подвижная фаза ацетонитрил-вода (77:23), скорость подвижной фазы 1,0 - 1,5 мл/мин., аликвота, вводимая в инжектор 10 - 15 мкл; чувствительность рефрактометрического детектора устанавливают таким образом, чтобы ввод 100 мкг фруктозы, глюкозы или сахарозы соответствовал отклонению пера на полную шкалу самописца при уровне шума от 1 до 3 % от полной шкалы; входное напряжение регистрирующего потенциометра (самописца) или интегратора 10 mV.
Ориентировочный порядок времен удерживания углеводов в минутах при использовании амино-фазы: фруктоза - 6±2, сорбит 6,5±2, глюкоза 7,5±1, сахароза 10±1.
Анализ ВЭЖХ с использованием колонки и предколонки с катионитом в форме Са++.
Условия ВЭЖХ: катионообменная колонка и предколонка «Rezex RCU-USP Sugar Alcohols» в форме Са++, температура колонки 80 - 90 °С, подвижная фаза бидистиллированная, деионизированная и фильтрованная вода, скорость подвижной фазы 0,4 - 0,8 мл/мин., аликвота, вводимая в инжектор 10 - 15 мкл; чувствительность хроматографа устанавливается таким образом, чтобы 30 мкг фруктозы, глюкозы и сорбита соответствовало отклонению пера на полную шкалу самописца при уровне шума от 1 до 3 % от полной шкалы; входное напряжение регистрирующего потенциометра (самописца) или интегратора 10 mV.
Ориентировочный порядок времен удерживания углеводов в минутах при использовании колонки и предколонки с катионитом в форме Са++: сахароза 10±1, глюкоза 12±0,5, фруктоза - 14,5±0,2, сорбит 22±0,5.
Предел обнаружения метода 0,02 - 0,05 %. Стандартное отклонение сходимости (Sr) при концентрации сахаров 10 % не превышает 0,05 - 0,07. Среднее значение открываемости от внесенного количества сахаров - 97 - 99 %.
Перед работой хроматограф калибруют, для чего в инжектор с помощью микрошприца вводят 0,002, 0,005 и 0,01 см3 стандартной смеси углеводов, что соответствует количествам 20, 50 и 100 мкг фруктозы, глюкозы и сахарозы (детектор RIDK 102) или 6, 15 и 30 мкг (детектор MERK L 7490). Для каждого количества углеводов определяют площадь пика на хроматограмме. Аналогичным образом проводят калибровку стандартного раствора сорбита.
Количественное определение углеводов
Идентификацию и количественное определение углеводов осуществляют методом внешних стандартов.
В инжектор хроматографа вводят 0,01 см3 (10 мкл) фильтрата. Определяют площадь пика углевода, соответствующего по времени удерживания одному из стандартов. Расчет концентрации фруктозы, глюкозы, сорбита и сахарозы проводят по формуле:
![]() ,
%, где
,
%, где
С - концентрация углевода в образце, %,
V1 - объем, до которого доведена навеска при разбавлении водой, см3 (100 см3),
V2 - объем фильтрата, внесенный в хроматограф, 0,01 см3,
m - масса стандарта, введенного в хроматограф, мкг,
М - навеска образца, взятая для анализа, г.
Если пик углевода в образце выходит за пределы шкалы самописца, анализ проводят повторно после разбавления фильтрата водой, т.е. после увеличения объема V1.
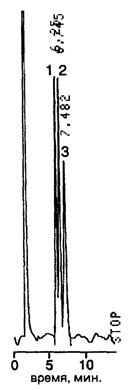
Рис. 6. Хроматограмма БАД на основе вишневого сока.
5. Определение массовой концентрации кофеина, теобромина, теофиллина
Принцип метода
Метод определения массовой концентрации кофеина, теобромина, теофиллина в пищевых продуктах, БАД и безалкогольных напитках основан на применении высокоэффективной жидкостной хроматографии (ВЭЖХ).
Метод анализа кофеина и теофиллина в БАДах включает следующие этапы:
· экстракцию пуриновых алкалоидов из БАД в дистиллированной воде при нагревании;
· фильтрование полученного раствора;
· обнаружение и количественное определение с помощью ВЭЖХ с УФ-детектором с использованием метода внешнего стандарта.
Метод анализа теобромина, теофиллина и кофеина в пищевых продуктах и БАД на основе какао включает следующие этапы:
· экстакцию в дистиллированной воде, рН 7,3 - 7,8 при нагревании;
· переэкстракцию в хлороформ;
· переэкстракцию в фосфатный буфер, рН 3,0
· фильтрование полученного раствора;
· обнаружение и количественное определение пуриновых алкалоидов с помощью ВЭЖХ с УФ-детектором с использованием метода внешнего стандарта.
Средства измерений, материалы и реактивы
Жидкостной хроматограф с насосом высокого давления с подачей растворителя от 0,1 до 5,0 см3/мин., оборудованный спектрофотометрическим детектором с переменной длиной волны и системой для сбора и обработки хроматографических данных Мультихром, версия 1,5х (Амперсенд, Россия).
Колонка и предколонка хроматографические с силикагелем, химически связанным с октадецилсиланом (силикагель С18) с размером частиц 5 мкм; длина колонки - 25 см, предколонки - 4,5 см, внутренний диаметр колонок - 0,46 см;
Микрошприцы МШ-10 и МШ-25 для жидкостной хроматографии.
Аппарат для встряхивания проб типа АВУ-6С, ТУ 64-1-2451-78.
Весы лабораторные общего назначения по ГОСТ 24104 с наибольшим пределом взвешивания 200 г и погрешностью ±0,0001 г.
Дистиллятор
рН-метр, модель «рН 150М».
Колбы мерные наливные 2-50-2, 2-100-2, 2-250-2, 1-1000-2 по ГОСТ 1770.
Пипетки 4-1-2 или 5-1-2, 4-2-10 или 5-2-10, 4-2-25 или 5-2-25 по ГОСТ 29227.
Термометр жидкостный стеклянный с пределами измерения 0 - 100 °С и ценой деления 1 °С по ГОСТ 28498.
Ацетонитрил, ч., ТУ 6-09-3534-74.
Бумага фильтровальная «красная лента» и «синяя лента» по ТУ 6-09-1678-95.
Баллон с сжатым азотом марки ПНГ;
Вода дистиллированная, ГОСТ 6709.
Калий фосфорнокислый однозамещенный, ч., ГОСТ 6552, раствор массовой концентрацией 2,72 г/дм3, доведенный до рН 3,2 добавлением 2 - 2,5 см3 ортофосфорной кислоты.
Кислота уксусная, ГОСТ 61-75, ч.д.а.
Кислота о-фосфорная, ч, ГОСТ 6552.
Кофеин, теобромин, теофиллин по действующей нормативно-технической документации.
Натрий гидроокись, х.ч., ГОСТ 4228, раствор массовой концентрации 0,4 г/дм3.
Натрий хлористый, х.ч., ГОСТ 4233.
Подготовка к проведению измерений
Приготовление рабочих растворов кофеина, теобромина теофиллина (400 мг/см3)
Навески массой по 100,0±0,1 мг помещают в мерные колбы вместимостью 250 см3, до половины заполненные дистиллированной водой. Перемешивают до полного растворения, затем доводят дистиллятом до метки и перемешивают.
Приготовление стандартных растворов кофеина, теобромина, теофиллина (0,004 мг/см3).
Отмеряют пипеткой по 2,0 см3 каждого рабочего раствора кофеина, теобромина, теофиллина концентрациями 400 мг/см3, переносят в мерные колбы вместимостью 250 см3, доводят до метки дистиллированной водой и перемешивают.
Приготовление подвижной фазы
Приготовление подвижной фазы для анализа кофеина
50 см3 ацетонитрила помещают в мерную колбу вместимостью 1000 см3, добавляют 10 см3 уксусной кислоты, доводят дистиллированной водой до метки и перемешивают. Полученный раствор фильтруют через фильтровальную бумагу «красная лента».
Приготовление подвижной фазы для анализа смеси алкалоидов
2,72 г калия фосфорнокислого однозамещенного помещают в мерную колбу вместимостью 1000 см3, растворяют в дистиллированной воде, перемешивают до полного растворения, доводят дистиллированной водой до метки. Полученный раствор ортофосфорной кислотой доводят до рН 3,2 (2 - 2,5 см3). 850 см3 полученного буферного раствора смешивают с 150 см3 ацетонитрила и полученный раствор фильтруют через фильтровальную бумагу «красная лента».
Подготовка образцов
Образцы тонизирующих напитков («Кола», тоники) подвергают по необходимости дегазации по ГОСТ 6687.2 при температуре не более 25 °С и отфильтровывают через бумажный фильтр. Аликвоту напитков (2 - 5 см3) помещают в мерную колбу вместимостью 250 см3, доводят дистиллированной водой до метки, тщательно перемешивают.
Навеску пищевого продукта (2 - 5 ±0,01 г) (например, чай, кофе), БАД на основе натуральных растительных компонентов гуараны, орехов колы, листьев матэ и др. (5 - 10 капсул или таблеток, растертых в ступке) помещают в мерную колбу вместимостью 250 см3, добавляют до половины объема подогретой до 70 - 80 °С дистиллированной воды, встряхивают на аппарате для встряхивания в течение 15 - 20 мин, охлаждают до комнатной температуры, доводят дистиллированной водой до метки и тщательно перемешивают. Фильтруют через бумажный фильтр с размером пор 0,5 мкм.
Навеску кондитерского изделия (10 - 20 ±0,01 г), содержащего какао-порошок (карамели, шоколад, ореховые пасты, глазурь и др.), измельченную на терке или в ступке, помещают в коническую колбу вместимостью 250 см3 и добавляют 100 см3 дистиллированной воды, подщелоченной 5 - 10 каплями раствора гидроокиси натрия (рН 7,5 - 8,0). Смесь подогревают до температуры 45 - 50 °С, перемешивают на аппарате для встряхивания 20 - 30 мин, фильтруют через бумажный фильтр «красная лента» и отбирают 80 см3 фильтрата. Если фильтрация затруднена, смесь центрифугируют при 2000 об/мин и также отбирают 80 см3. К фильтрату добавляют 1,5 - 2 г хлористого натрия, перемешивают до растворения соли и переносят в делительную воронку. Пуриновые алкалоиды из водного раствора экстрагируют 50 см3 хлороформа. Хлороформный раствор отделяют и встряхивают с 50 см3 раствора калия фосфорнокислого однозамещенного, рН 3,2, после чего водный экстракт фильтруют через бумажный складчатый фильтр «синяя лента». Остатки хлороформа из фильтрата удаляют в токе азота. Аликвоту полученного экстракта вводят в хроматограф. При анализе используют метод внешнего стандарта.
Проведение анализа ВЭЖХ и расчет концентрации пуриновых оснований
Условия ВЭЖХ для хроматографического анализа кофеина: подвижная фаза ацетонитрил - вода дистиллированная - уксусная кислота (15:84:1). Скорость потока 0,7 см3/мин. Обнаружение по поглощению в УФ-области спектра при длине волны 272 нм, шкала чувствительности 0,01 Е. О. П. Вводимый объем образца 10,0 мм3.
Условия ВЭЖХ для хроматографического анализа смеси алкалоидов: подвижная фаза: раствор калия фосфорнокислого однозамещенного, рН 3,2-ацетонитрил (85 - 15), скорость потока 0,8 см3/мин, УФ-детектирование, длина волны 272 нм, шкала чувствительности 0,01 Е. О. П. Вводимый объем образца 10,0 мм3.
Примерные времена удерживания пуриновых алкалоидов: теобромин 4,9±0,1, теофиллин 7,7±0,1, кофеин 12,0±0,1.
Расчет концентраций пуриновых оснований
Массовую концентрацию теобромина, теофиллина или кофеина рассчитывают по формуле:
![]() ,
мг/кг, где
,
мг/кг, где
Sобp. - площадь пика исследуемого компонента;
Sст. - - площадь пика соответствующего стандарта;
V1 - объем анализируемого раствора пробы, мм3;
V2 - объем экстракта, введенного в хроматограф, мм3;
mcm. - масса стандарта пуринового алкалоида, введенная в хроматограф, нг;
Мобр. - навеска образца, взятая для анализа и соответствующая объему фильтрата после экстракции подщелоченной водой, г, (8 г).
Вычисления проводят до второго десятичного знака. За результат принимают среднее арифметическое результатов двух параллельных измерений и выражают целым числом с одним десятичным знаком.
Относительное допустимое расхождение между результатами двух параллельных определений при доверительной вероятности Р = 0,95 не должно превышать 13 %.
Характеристика погрешности измерений
При соблюдении всех регламентируемых методикой условий проведения измерений относительная характеристика погрешности (стандартное отклонение сходимости) результата анализа при концентрации кофеина в БАД 2,0 мг/таблетку или капсулу не превышает 0,154 (3,43 %), при концентрации теобромина в пищевом продукте 5 мг/100 г - 0,066 (4,89 %).
Предел обнаружения метода определения пуриновых алкалоидов (теобромина, теофиллина, кофеина) в пищевых продуктах составляет 1,0 - 2,0 мг/кг. Диапазон определяемых концентраций от 1,0 до 1000 мг/кг.
Среднее значение открываемости пуриновых алкалоидов в БАД при концентрациях от 2,0 до 20 мг/таблетку составляет 87 - 95 %, в пищевых продуктах при концентрациях от 5,0 до 25 мг/100 г - 82 - 90 %.
6. Определение массовой концентрации хинина
Приготовление стандартных растворов
Приготовление градуировочного раствора хинина (800 мг/см3)
Навеску хинина массой 200,0 мг помещают в мерную колбу вместимостью 250 см3, до половины заполненную бидистиллированной водой. Перемешивают до полного растворения, затем доводят бидистиллятом до метки и перемешивают.
Приготовление градуировочного раствора хинина (0,008 мг/см3).
Отмеряют пипеткой 2,0 см3 градуировочного раствора хинина концентрации 800 мг/см3, переносят в мерную колбу вместимостью 100 см3, доводят до метки бидистиллированной водой и перемешивают.
Приготовление подвижной фазы
2,72 г калия фосфорнокислого однозамещенного (KH2PO4) помещают в мерную колбу вместимостью 1000 см3, растворяют в бидистиллированной воде, перемешивают до полного растворения. Полученный раствор ортофосфорной кислотой (Н3РO4) доводят до рН 3,0 и фильтруют через фильтровальную бумагу с диаметром пор 0,5 мкм. 760 см3 полученного буферного раствора смешивают с 240 см3 ацетонитрила (С2Н3N). Срок годности полученного раствора не более недели.
Подготовка образца
Образцы напитков подвергают дегазации по ГОСТ 6687.2 при температуре не более 25 °С и отфильтровывают через бумажный фильтр.
Проведение анализа
Условия хроматографического анализа
Подвижная фаза 24 % ацетонитрила, 76 % 0,02 М раствора KH2PO4, доведенного ортофосфорной кислотой до рН 3,0. Скорость потока 2,0 мл/мин. УФ-детектирование, длина волны 254 нм.
Массовую концентрацию хинина рассчитывают сравнением площадей пиков образца и стандарта хинина по аналогии с формулой в разделе 5.
7. Определение содержания коэнзима Q10
Метод основан на определении высокоэффективной жидкостной хроматографией.
Оборудование и реактивы
Жидкостной хроматограф с УФ-детектором.
Стандартный раствор Коэнзим Q10 концентрацией 100 мкг/мл готовят в смеси этанол-эфир (1:1).
Подготовка проб
Коэнзим Q10 - убихинон, не растворим в воде, но хорошо растворим в спирте и эфире.
Из образца (обычно это капсулы) его выделяют растворением в смеси 96 %-ный этанол-эфир (1:1) Капсулы прокалывают в нескольких местах иглой, заливают смесью растворителей и оставляют при комнатной температуре в темном месте в плотно укупоренной склянке на сутки. Через сутки пробы хроматографируют.
Проведение испытания
Условия ВЭЖХ: Колонка из нержавеющей стали, длина 150 мм, диаметр 4 - 6 мм. Сорбент - Сепарон SGX С18, размер частиц 7 мкм. Перед работой новую колонку промывают 40 мл изопропанола, затем 80 мл деионизированной воды, после чего уравновешивают колонку подвижной фазой до стабильной нулевой линии.
Подвижная фаза: 96 % этанол, очищенный перегонкой. Детектирование: спектрофотометр, длина волны УФ 290 нм, шкала чувствительности 0,05 AUFS. скорость потока 10 мл/мин, время удерживания коэнзима Q - 8,2 мин.
Обработка результатов: стандартный образец и испытываемую пробу хроматографируют не менее трех раз. Расчет содержания Коэнзима Q производят методом абсолютной калибровки. Ошибка метода ±5 %.
8. Определение L-карнитина (g-триметил-р-гидроксибутиробетаин)
L-карнитин - гигроскопические кристаллы, хорошо растворимые в воде, в этаноле. Мало растворим в ацетоне, изопропаноле, не растворим в эфире.
Метод определения массовой концентрации L-карнитина в БАД и пищевых продуктах основан на применении высокоэффективной жидкостной хроматографии.
Метод анализа включает следующие этапы:
· экстракцию L-карнитина из БАД и пищевых продуктов подщелоченной дистиллированной водой при нагревании;
· осаждение высокомолекулярных составляющих экстракта ацетатом цинка;
· фильтрование полученного раствора;
· обнаружение и количественное определение с помощью йон-парной ВЭЖХ с УФ детектором и использованием метода внешнего стандарта.
Средства измерений, материалы и реактивы
Жидкостной хроматограф с насосом высокого давления с подачей растворителя от 0,1 до 5,0 см3/мин., оборудованный спектрофотометрическим детектором с переменной длиной волны и системой для сбора и обработки хроматографических данных Мультихром, версия 1,5х (Амперсенд, Россия).
Колонка и предколонка хроматографические с силикагелем, химически связанным с октадецилсиланом (силикагель С18) с размером частиц 5 мкм; длина колонки - 25 см, предколонки - 4,5 см, внутренний диаметр колонок - 0,46 см;
Микрошприцы МШ-10 и МШ-25 для жидкостной хроматографии.
Аппарат для встряхивания проб типа АВУ-6С, ТУ 64-1-2451-78.
Весы лабораторные общего назначения по ГОСТ 24104 с наибольшим пределом взвешивания 200 г и погрешностью ±0,0001 г.
Дистиллятор
рН-метр, модель «рН 150М».
Колбы мерные наливные 2-50-2, 2-100-2, 2-250-2, 1-1000-2 по ГОСТ 1770.
Пипетки 4-1-2 или 5-1-2, 4-2-10 или 5-2-10, 4-2-25 или 5-2-25 по ГОСТ 29227.
Термометр жидкостный стеклянный с пределами измерения 0 - 100 °С и ценой деления ГС по ГОСТ 28498.
Ацетонитрил, ч., ТУ 6-09-3534-74.
Бумага фильтровальная «красная лента» и «синяя лента» по ТУ 6-09-1678-95.
Баллон со сжатым азотом марки ПНГ;
Вода дистиллированная, ГОСТ 6709.
Кислота о-фосфорная, ч, ГОСТ 6552.
Додецилсульфат, натриевая соль, SERVA, раствор 0,005М в дистиллированной воде.
L-карнитин по действующей нормативно-технической документации.
Натрий гидроокись, х.ч., ГОСТ 4228, раствор массовой концентрации 0,4 г/дм3.
Натрий хлористый, х.ч., ГОСТ 4233.
Цинк уксуснокислый 2-водный, ч.д.а., ГОСТ 5823-78, раствор 30 %-ный в дистиллированной воде.
Подготовка к проведению измерений
Приготовление рабочего раствора L-карнитина (400 мг/см3)
Навеску L-карнитина массой 100,0±0,1 мг помещают в мерную колбу вместимостью 250 см3, до половины заполненную дистиллированной водой. Перемешивают до полного растворения, затем доводят дистиллятом до метки и перемешивают.
Приготовление стандартного раствора L-карнитина (0,004 мг/см3).
Отмеряют пипеткой 2,0 см3 рабочего раствора L-карнитина концентрацией 400 мг/см3, переносят в мерную колбу вместимостью 200 см3, доводят до метки дистиллированной водой и перемешивают.
Приготовление подвижной фазы
1,44 г додецилсульфата натрия помещают в мерную колбу вместимостью 1000 см3, растворяют в дистиллированной воде, перемешивают до полного растворения, доводят дистиллированной водой до метки. Полученный раствор ортофосфорной кислотой доводят до рН 2,5 (2,5 - 3,06 см3). 850 см3 полученного буферного раствора смешивают с 350 см3 ацетонитрила и полученный раствор фильтруют через фильтровальную бумагу «красная лента».
Подготовка проб
Выделение L-карнитина осуществляли следующим образом: навеску (2 г) измельченного БАД или продукта помещали в мерную колбу вместимостью 250 см3, добавляли 125 см3 горячей (60 - 65 °С) дистиллированной воды, подщелоченной раствором NaOH массовой концентрации 0,4 г/дм3 (рН 8,0 - 8,5), встряхивали на аппарате для встряхивания 30 минут, охлаждали до комнатной температуры, добавляли 30 см3 30 %-ного раствора ацетата цинка доводили дистиллированной водой до метки, тщательно перемешивали и фильтровали через бумажный фильтр «красная лента» или через капроновый фильтр марки 0,45 мкм RC.
Проведение испытания
Условия ВЭЖХ: подвижная фаза раствор 0,005 М додецилсульфоната натрия - ацетонитрил (65:35). Скорость потока 1,0 см3/мин. Обнаружение по поглощению в УФ-области спектра при длине волны 210 нм, шкала чувствительности 0,01 Е.О.П. Вводимый объем образца 10,0 мм3. Время удерживания карнитина - 7,5±1,0 мин.
Расчет концентраций L-карнитина
Массовую концентрацию L-карнитина рассчитывают по формуле:
![]() ,
мг/100 г, где
,
мг/100 г, где
![]() - площадь пика исследуемого компонента;
- площадь пика исследуемого компонента;
![]() - площадь пика стандарта;
- площадь пика стандарта;
V1 - объем анализируемого раствора пробы, мм3;
V2 - объем экстракта, введенного в хроматограф, мм3;
![]() - масса стандарта, введенная в хроматограф, нг;
- масса стандарта, введенная в хроматограф, нг;
![]() - навеска образца, взятая для анализа.
- навеска образца, взятая для анализа.
Обработка результатов. Характеристика погрешности измерений
За результат принимают среднее арифметическое результатов трех параллельных измерений и выражают целым числом с одним десятичным знаком. Относительное допустимое расхождение между результатами параллельных определений при доверительной вероятности Р = 0,95 не должно превышать 13 %. Ошибка метода ±5 %.
Среднее значение открываемости L-карнитина в БАД при концентрациях 10 - 20 мг/таблетку или в пищевых продуктах при концентрациях от 100 мг % составляет - 85 - 93 %.
9. Определение полифенольных соединений
Принцип метода
Суммарное содержание фенольных соединений определяют модифицированным методом Фолина-Чокальтеу. Полифенольные соединения окисляются реактивом Фолина-Чокальтеу, состоящим из смеси фосфорно-вольфрамовой H2PW12O40 и фосфорно-молибденовой Н3РMo12О40 кислот, который, в свою очередь, восстанавливается в смесь окислов вольфрама (W8O23) голубого цвета и молибдена (Mo8O23). Абсорбция раствора при 750 нм пропорциональна содержанию фенольных соединений. В качестве полифенольного стандарта используют галловую кислоту.
Приборы и реактивы
Спектрофотометр СФ-26, СФ-46 или подобный, позволяющий проводить измерения при длинах волн 200 - 350 нм, с допустимой абсолютной погрешностью измерений коэффициента пропускания не более 1 %.
Колориметр фотоэлектрический лабораторный КФК-2 (или аналогичный) ГОСТ 12083.
Аппарат для встряхивания проб типа АВУ-6С, ТУ 64-1-2451-78.
Весы лабораторные общего назначения по ГОСТ 24104 с пределом взвешивания 200 г и погрешностью ±0,0001 г.
рН-метр лабораторный ЭВ-74 (или аналогичный) по ТУ 25-05-27-57.
Дистиллятор.
Вода дистиллированная по ГОСТ 6709.
Колбы мерные наливные 2-50-2, 2-100-2, 2-250-2, 1-1000-2 по ГОСТ 1770.
Пипетки 4-1-2 или 5-1-2, 4-2-10 или 5-2-10, 4-2-25 или 5-2-25 по ГОСТ 29227.
Стакан мерный В-1-500ТС вместимостью 500 см3 по ГОСТ 25336.
Цилиндры 2-1000 и 2-100 по ГОСТ 1770.
Натрия вольфрамат по ГОСТ 18289.
Натрия молибдат по ГОСТ 10931.
Кислота ортофосфорная 85 %-ная по ГОСТ 6552.
Кислота соляная конц. по ГОСТ 3118.
Литий сернокислый (сульфат лития).
Натрий углекислый (карбонат натрия) по ГОСТ 83.
Водорода перекись по ГОСТ 177.
Бром по ГОСТ 4109.
Спирт этиловый ректификованный по ГОСТ 5962.
Галловая кислота по действующей нормативно-технической документации.
Подготовка к выполнению измерений
Приготовление реактива Фолина
100 г вольфрамовокислого натрия и 25 г молибденовокислого натрия растворяют в 700 см3 дистиллированной воды. Добавляют 50 см3 85 %-ной фосфорной кислоты (d = 1,71 г/см3), 100 см3 концентрированной соляной кислоты (d = 1,19 г/см3) и кипятят на водяной бане в колбе с обратным холодильником под тягой в течение 10 ч. Добавляют 150 г сернокислого лития, несколько капель брома и снова кипятят в течение 15 мин. Смесь охлаждают до комнатной температуры, переносят в мерную колбу вместимостью 1 дм3 и доводят объем до метки дистиллированной водой. Хранят реактив в темной бутылке со шлифом в холодильнике.
Приготовление раствора карбоната натрия
В мерную колбу вместимостью 1 дм3 наливают около 500 см3 дистиллированной воды и растворяют 200 г карбоната натрия при встряхивании. Доводят до метки дистиллированной водой и перемешивают. Получают раствор карбоната натрия с концентрацией 20 %.
Раствор галловой кислоты
В мерный стакан вместимостью 500 см3 помещают 50 см3 этилового спирта, доводят до метки 400 см3 дистиллированной водой. Погружают в стакан электроды рН-метра и добавляют соляную кислоту до значения рН 3,2 - 3,25. Переносят полученный раствор в мерную колбу вместимостью 500 см3, при встряхивании растворяют 15 мг галловой кислоты и доводят дистиллированной водой до метки. Концентрация галловой кислоты 0,03 мг/см3.
Построение градуировочной кривой
1, 2, 5, 10, 20 см3 раствора галловой кислоты помещают в 5 мерных колб вместимостью 100 см3. В шестую колбу вносят 1 см3 дистиллированной воды - контрольный раствор.
В каждую мерную колбу добавляют по 1 см3 реактива Фолина-Чокальтеу, по 10 см3 раствора карбоната натрия, доводят дистиллированной водой до метки и перемешивают.
Через 20 - 30 мин измеряют оптическую плотность растворов в кювете 10 мм при длине волны 750 нм против контрольного раствора.
По полученным данным строят градуировочную кривую, откладывая по оси абсцисс массовую концентрацию галловой кислоты, а по оси ординат - оптическую плотность.
Выполнение измерений
БАД на растительной и фруктовой основе (таблетки, капсулы) растирают в ступке, навеску 1 - 2 г помещают в мерную колбу вместимостью 100 см3, заполненную до половины дистиллированной водой, нагретой до 40 °С, и встряхивают на аппарате для встряхивания в течение 45 - 60 мин, периодически подогревая колбу.
Сокосодержащие БАД (растворы, сиропы) разбавляют в 5 - 10 раз дистиллированной водой.
1 см3 полученных растворов помещают в мерные колбы вместимостью 100 см3 и далее выполняют действия как в разделе «Построение градуировочной кривой».
Вычисление результатов определения.
Значение массовой концентрации фенольных веществ (С) в мг/дм3 по галловой кислоте определяют по градуировочной кривой. Полученную величину умножают на коэффициент разведения - 100, если экстракт разбавляли в 5 раз или вычисляют по формуле:
С = А´ОП, где
А - коэффициент разбавления,
ОП - оптическая плотность.
Вычисления проводят до первого десятичного знака и округляют до целого числа. За окончательный результат принимают среднее арифметическое результатов двух параллельных определений.
Характеристика погрешности измерений
Допустимое значение погрешности измерений массовой концентрации фенольных соединений в БАД на растительной основе или сокосодержащих БАД не должно превышать:
для диапазона концентраций в экстракте или растворе 100 - 600 мг/дм3 - ±8 мг/дм3,
для диапазона концентраций в экстракте или растворе 600 - 3000 мг/дм3 - ±33 мг/дм3.
10. Определение флавоноидов
10.1. Введение
Флавоноиды представляют собой большую группу природных полифенольных соединений с двумя ароматическими кольцами, имеющими основную структуру С6-С3-С6. Флавоноиды являются мощными природными антиоксидантами, обладающими широким спектром биологической активности. Особенно богаты флавоноидами высшие растения, относящиеся к семействам розоцветных (различные виды боярышников, черноплодная рябина), бобовых (софора японская, стальник полевой, солодка), гречишных (различные виды горцев - перечный, почечуйный, птичий: гречиха), астровых (бессмертник песчаный, сушеница топяная, пижма), яснотковых (пустырник сердечный) и др.
В основе структуры большинства флавоноидов лежат производные флавана (2-фенил-хроман или 2-фенил-бенз-g-пиран), приведенного на рис. 7, которые, в свою очередь, разделяются на производные хромана и хромона.
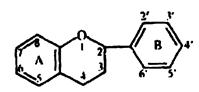
Рис. 7.
К производным хромана относятся катехины, лейкоантоцианидины и антоцианидины.
Катехины. Основные структуры катехинов (флаван-3-олов) приведены на рис. 8.
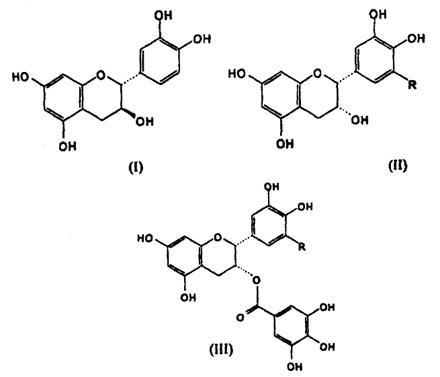
Рис. 8. Структуры основных катехинов (флаван-3-олов):
(I) - (+)-катехин; (II) - R = Н (-)-эпикатехин, R = ОН - (-)-эпигаллокатехин; (III) - R = Н (-)-эпикатехингаллат; R = ОН (-)-эпигаллокатехингаллат.
Лейкоантоцианидины. Как видно из приведенной структуры на рис. 9 лейкоантоцианидины или проантоцианидины (флаван-3,4-диолы) представляют собой 4-гидроксипроизводное катехина.
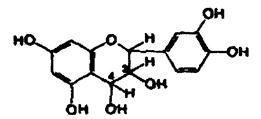
Рис. 9.
Антоцианидины. Особенность строения антоцианидинов (рис. 10) в том, что кислород в пирановом кольце в кислой среде образует оксониевый катион. Поэтому антоцианидины в кислом растворе ведут себя как катионы и образуют соли с кислотами, а в щелочном растворе - как анионы и образуют соли с основаниями. Антоцианидины являются интенсивно окрашенными природными пигментами, их окраска изменяется в зависимости от рН среды. Оксониевые формы (соли катионов) антоцианидинов окрашены в красный цвет с оттенками: желтоватыми (пеларгонидин), фиолетовым (цианидин), синеватым (дельфинидин). Щелочные соли окрашены в синий цвет.
Антоцианидины являются агликонами большого класса природных красителей -антоцианинов (см. раздел 1).
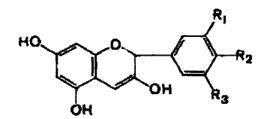
Рис. 10.
Среди других представителей этой группы следует отметить 2-фенилпроизводные хромана: флаваноны и флаванонолы, структуры которых представлены на рис. 11.
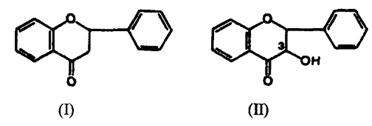
Рис. 11.
(I) - флаванон, (II) - флаванонол.
К флаванонам относятся нарингенин - 5,7,4¢-тригидроксифлаванон, гесперетин - 5,7,3¢-тригидрокси-4¢-метоксифлаванон и эриодиктиол - 5,7,3,4-тетрагидро-ксифлаванон. Наиболее распространенными в экстрактах цитрусовых являются 7-O-гликозил-нарингенин - нарингин и 7-О-гликозилгесперетин - геспередин (см. раздел 11.2).
В БАД на основе экстрактов лиственницы и ряда других растений содержатся производные флаванон-3-олов: дигидрокверцетин - 5,7,3,4-тетрагидроксифлаванон-3-ол и дигидрокемпферол - 5,7,4-тригидроксифлаванон-3-ол (см. раздел 11.3).
Из производных хромона, представленных на рис. 12, наиболее распространены в растениях соединения типа флавона и флавонола, имеющих непредельную связь в положении 2, 3. В отличии от их гидрированных аналогов - флаванонов и флаванонолов - это наиболее устойчивые флавоноиды.
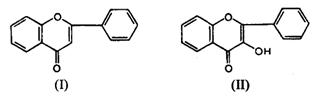
Рис. 12.
(I) - флавон, (II) - флавонол.
Флавоноиды, у которых фенильная группа с С2 смещена к С3, называются изофлавоноидами (рис. 13).
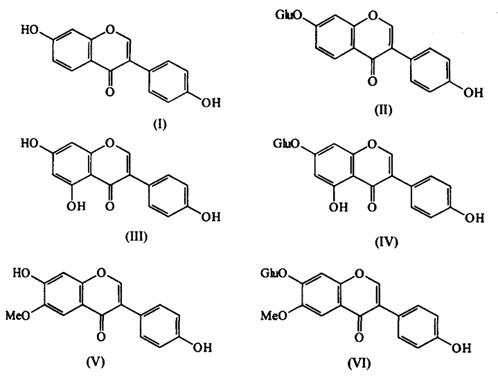
Рис. 13.
(I) - даидзеин, (II) - даидзин, (III) - генистеин, (IV) - генистин, (V) - глицитеин, (VI) - глицитин.
Структуры и наименования основных флавонолов представлены на рис. 14, структуры флавонов - на рис. 15.
10.2. Методы определения флавоноидов
В основе количественного определения флавоноидов лежит спектрофотометрия при длине волны 415 нм продуктов взаимодействия с раствором хлорида алюминия. К флавоноидам относятся производные флавона: флавонолы (рутин - рутинозид кверцетина, гиперозид - галактозид кверцетина, морин, кверцетин, мирицетин, кемпферол, кверцитрин, галангин) и флавоны (хризин, апигенин, лютеолин, изовитексин, изоориентин). Для определения производных флавана: флаванон-3-олов (дигидрокверцетин или таксифолин, дигидрокемферол, силибин), флаванонов (нарингин, ±-нарингенин, гесперетин) используется спектрофотометрия при длине волны 495 нм продуктов их реакции с 2,4-динитрофенилгидразином.
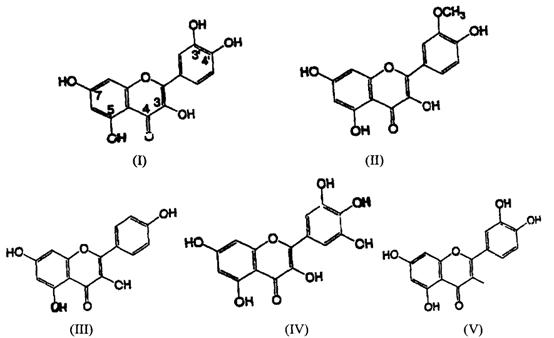
Рис. 14.
(I) - кверцетин, (II) - изорамнетин, (III) - кемпферол, (IV) - мирицитин, (V) - рутин.
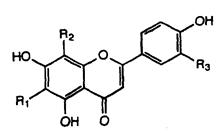
|
Флавон |
r1 |
R2 |
R3 |
|
Апигенин |
Н |
Н |
Н |
|
Лютеолин |
Н |
Н |
ОН |
|
Изовитексин |
Глюкозил- |
Н |
Н |
|
Витексин |
Н |
Глюкозил- |
Н |
|
Изоориентин |
Глюкозил- |
Н |
ОН |
|
Ориентин |
Н |
Глюкозил- |
ОН |
Рис. 15.
Принцип колориметрического метода, основанного на взаимодействии с алюминий хлоридом, заключается в том, что реагент образует кислотоустойчивые комплексы с С-4 кето группой и или с С-3 или С-5 гидроксильной группой флавонов и флавонолов, имеющие максимумы поглощения в диапазоне длин волн 415 - 440 нм.
Принцип взаимодействия с 2,4-динитрофенилгидразином основан на его реакции с кетонами и альдегидами с образованием 2,4-динитрофенилгидразонов. Следовательно, флавоны, флавонолы и изофлавоны с С2 - С3 двойной связью не способны реагировать с 2,4-динитрофенилгидразином, в то время, как гидразоны и флавононы образуют соединения, имеющие максимум поглощения при 495 нм.
Приборы и реактивы
Спектрофотометр СФ-26, СФ-46 или подобный (Shimadzu UV-160A, Япония), позволяющий проводить измерения при длинах волн 190 - 900 нм, с допустимой абсолютной погрешностью измерений коэффициента пропускания не более 1 %;
Метанол - яд для жидкостной хроматографии, ос.ч. по ТУ 6-09-14-2192-85;
Спирт этиловый ректификованный очищенный дистилляцией.
Алюминия хлористый, 6-водный, х.ч., ГОСТ 3759-75, раствор 10 %-ный в 95 %-ном этаноле;
2,4-динитрофенилгидразин (2,4-Д), раствор: 1 г 2,4-Д растворяют в 2 см3 96 %-ной серной кислоты и доводят метанолом до объема 100 см3, концентрация полученного раствора 10 мг/см3;
Натрия гидроокись, х.ч.: 1 %-ный раствор в 70 %-ном метаноле;
Натрия ацетат, 1 М раствор.
Приготовление стандартных растворов для калибровки спектрофотометра
10±0,1 мг кверцетина растворяют в 80 %-ном этаноле и затем разбавляют в мерных колбах до концентраций 25, 50 и 100 мкг/см3.
20±0,1 мг ±-нарингенина растворяют в метаноле и затем разбавляют до концентрации 500, 1000 и 2000 мкг/см3.
Подготовка образца
1,0 г измельченного образца кипятят в 30 см3 90 %-ного этилового спирта, содержащего 0,5 % концентрированной серной кислоты, на кипящей водяной бане с обратным холодильником в течение 30 мин. Надосадочную жидкость сливают в мерную колбу вместимостью 100 см3 и экстракцию повторяют еще раз. Объединенную смесь охлаждают, фильтруют и доводят объем до 100 см3 90 %-ным этанолом (раствор А).
Колориметрический метод с алюминий хлоридом (суммарное определение флавонолов)
По 0,5 см3 каждого разбавленного стандарта кверцетина смешивают с 1,5 см3 95 %-ного этанола, 0,1 см3 10 %-ного хлорида алюминия, 0,1 см3 1 М ацетата натрия и 2,8 см3 дистиллированной воды. Смесь инкубируют при комнатной температуре 30 мин и измеряют оптическую плотность полученного раствора при 415 нм. Для приготовления раствора сравнения используют разбавленный в дистиллированной воде 10 %-ный хлорид алюминия.
Колориметрический метод с 2,4-динитрофенилгидразином (суммарное определение флаванонов)
По 1,0 см3 каждого разбавленного стандартного раствора ±-нарингенина смешивают с 2,0 см3 1 %-ого раствора 2,4-Д и 2,0 см3 метанола. Смесь выдерживают при 50 °С в течение 50 мин. После охлаждения до комнатной температуры к смеси добавляют 5 см3 1 %-ного раствора едкого натра в 70 %-ном метаноле и инкубируют при комнатной температуре в течение 2-х минут. Затем к 1 см3 смеси добавляют 5 см3 метанола и центрифугируют при 1000 об/мин, в течение 10 мин. Отделяют супернатант и в мерной колбе доводят до объема 25 см3. Измеряют оптическую плотность раствора при 495 нм против раствора сравнения - метанола.
Проведение испытаний
Отбирают по 0,5 см3 гидролизата (раствор А) и проводят колориметрические реакции с хлоридом алюминия и 2,4-Д как описано выше. Определяют содержание флавоноидов согласно калибровочным графикам.
11. Анализ индикаторных показателей некоторых БАД на растительной основе
11.1. Определение катехинов и галловой кислоты в БАД на основе зеленого чая и в травяных чаях
Катехины - это флаван-3-олы, структура которых представлены на рис. 8.
Приборы и реактивы
Жидкостной хроматограф с насосом высокого давления с подачей растворителя от 0,1 до 5,0 см3/мин., обеспечивающий работу в режиме градиентного элюирования, оборудованный спектрофотометрическим детектором с переменной длиной волны и системой для сбора и обработки хроматографических данных Мультихром, версия 1,5х (Амперсенд, Россия).
Колонка и предколонка хроматографические с силикагелем, химически связанным с октадецилсиланом (силикагель С18) с размером частиц 5 мкм; длина колонки - 15 см, предколонки - 4,5 см, внутренний диаметр колонок - 0,46 см;
Микрошприцы МШ-10 и МШ-25 для жидкостной хроматографии.
Колбы мерные наливные 2-50-2, 2-100-2, 2-250-2, 1-1000-2 по ГОСТ 1770.
Пипетки 4-1-2 или 5-1-2, 4-2-10 или 5-2-10, 4-2-25 или 5-2-25 по ГОСТ 29227.
Ацетонитрил, ч., ТУ 6-09-3534-74.
Спирт этиловый ректификованный.
Кислота о-фосфорная, ч, ГОСТ 6552, раствор 0,1 %: 1,2 см3 фосфорной кислоты помещают в мерную колбу вместимостью 1 дм3, добавляют около 950 см дистиллированной воды, перемешивают, охлаждают до комнатной температуры, доводят до метки и перемешивают.
Метанол - яд для жидкостной хроматографии, ос.ч. по ТУ 6-09-14-2192-85;
Спирт этиловый ректификованный очищенный дистилляцией.
Капроновый фильтр марки 0,45 мкм RC;
Фумажные фильтры обеззоленные марки ФОМ.
(+)-Катехин, (+)-эпикатехин, (-)-эпигаллокатехин, (-)-эпигаллокатехин галлат, галловая кислота - SIGMA Chemical Co.
Приготовление стандартного раствора
5±0,1 мг (+)-катехина предварительно высушенного при 105 °С в течение 1 ч, помещают в мерную колбу вместимостью 100 см3, добавляют около 70 см3 0,1 %-ного раствора фосфорной кислоты, перемешивают до полного растворения вещества, охлаждают до комнатной температуры, доводят до метки 0,1 %-ным раствором фосфорной кислоты и перемешивают. Концентрация полученного стандартного раствора катехина 0,05 мг/см3. 5 см3 полученного раствора переносят в мерную колбу вместимостью 50 см3, доводят до метки 0,1 %-ным раствором фосфорной кислоты и перемешивают. Концентрация полученного рабочего стандартного раствора катехина 0,005 мг/см3. В хроматограф вводят 10 мкл.
Подготовка образца
1 г порошка экстракта листьев, 2 г измельченных листьев, таблеток или содержимого капсул помещают в химический стакан вместимостью 250 см3, добавляют 50 см3 0,1 %-ного раствора фосфорной кислоты и проводят экстракцию в ультразвуковой бане в течение 5 мин. Полученный раствор фильтруют через бумажный фильтр «синяя лента» в мерную колбу вместимостью 250 см3 или при необходимости центрифугируют при 3000 об/мин 5 мин, а затем супернатант помещают в мерную колбу на 250 см3, доводят до метки 0,1 %-ным раствором фосфорной кислоты и перемешивают (раствор А). Раствор анализируют с помощью ВЭЖХ.
Проведение анализа ВЭЖХ
Условия хроматографического анализа: колонка Hypersil ODS, размер колонки 250´4,6 мм, размер частиц 5 мкм, подвижная фаза ацетонитрил- 85 %-ная фосфорная кислота (15:85), скорость подвижной фазы 1,0 см3/мин., объем петли инжектора 20 мм3. УФ детектор, длина волны 280 нм. Времена удерживания: галловая кислота 2,9±0,1 мин, эпигаллокатехин 4,5±0,1 мин, катехин 5,8±0,2 мин, эпикатехин 6,8±0,2 мин., эпигаллокатехин галлат 9,8±0,2 мин., галлокатехин галлат 12,0±0,1 мин, эпикатехин галлат 13,5±0,1 мин, катехин галлат 15,5±0,1 мин.
Количественный расчет
При количественном анализе используют метод абсолютной калибровки.
Количественный расчет каждого катехина в пересчете на катехин проводят по формуле:
![]() %,
где
%,
где
![]() - площадь пика исследуемого компонента;
- площадь пика исследуемого компонента;
![]() - площадь пика стандарта катехина;
- площадь пика стандарта катехина;
V1 - объем анализируемого раствора пробы, мм3;
V2 - объем экстракта, введенного в хроматограф, мм3;
![]() - масса стандарта катехина, введенная в
хроматограф, мг;
- масса стандарта катехина, введенная в
хроматограф, мг;
![]() - навеска образца, взятая для анализа, мг,
- навеска образца, взятая для анализа, мг,
К - коэффициент пересчета содержания индивидуального катехина относительно катехина - согласно нижеприведенной таблице:
|
Катехин |
К, по катехину |
|
(+)-катехин |
1,000 |
|
(-)-эпикатехин |
1,020 |
|
(-)-катехин галлат |
0,327 |
|
(-)-эпикатехин галлат |
0,382 |
|
(-)-галлокатехин галлат |
0,482 |
|
(-)-эпигаллокатехин галлат |
0,543 |
|
(-)-эпигаллокатехин |
3,450 |
Относительное стандартное отклонение, вычисленное по площадям пиков не менее двух параллельных измерений катехина не должно превышать 0,4 %.
11.2. Определение флаванонов в БАД на фруктовой основе
Гесперидин, нарингенин относятся к разряду флаванглюкозидов, широко представленных во фруктах, особенно в кожуре зеленых апельсинов. При отжатии сока под высоким давлением флаваноны переходят в сок.
Приборы и реактивы
Жидкостной хроматограф с насосом высокого давления с подачей растворителя от 0,1 до 5,0 см3/мин., обеспечивающий работу в режиме градиентного элюирования, оборудованный спектрофотометрическим детектором с переменной длиной волны и системой для сбора и обработки хроматографических данных Мультихром, версия 1,5х (Амперсенд, Россия).
Колонка и предколонка хроматографические с силикагелем, химически связанным с октадецилсиланом (силикагель С18) с размером частиц 5 мкм; длина колонки - 15 см, предколонки - 4,5 см, внутренний диаметр колонок - 0,46 см.
Микрошприцы МШ-10 и МШ-25 для жидкостной хроматографии.
Кислота серная, х.ч.
Ацетонитрил, ч., ТУ 6-09-3534-74.
Спирт этиловый ректификованный.
Фумажные фильтры обеззоленные марки ФОМ.
Гесперидин, раствор в 50 %-ном этиловом спирте концентрацией 0,02 мг/см3.
Нарингенин, раствор в 50 %-ном этиловом спирте концентрацией 0,02 мг/см3.
Подготовка образца
1 г измельченных таблеток или содержимого капсул помещают в плоскодонную колбу, добавляют 40 см3 50 % этилового спирта, смесь нагревают на водяной бане при 55 - 60 °С в течение 30 мин. Экстракцию повторяют пятикратно. Спиртовые экстракты, охлаждают, фильтруют, доводят объем до 250 см3 50 %-ным этиловым спиртом.
Проведение анализа
Условия хроматографического разделения: колонка ODS-Hypersil (HP 799160D-552), размеры колонки 100´2,1 мм, размер частиц 5 мкм, объем петли инжектора 20 мм3. Подвижная фаза: раствор серной кислоты, рН 2,5 - ацетонитрил (85:15), скорость потока 0,5 см3/мин., УФ детектор, длина волны 280 нм. Ориентировочные времена удерживания нагингенина 4,8±0,1 мин., гисперидина 6,0±0,1 мин.
При количественном анализе используют метод абсолютной калибровки. Относительное стандартное отклонение, вычисленное по площадям пиков не менее двух параллельных измерений стандартов гесперидина и нарингенина, не должно превышать 0,4 %.
11.3. Определение флаванонолов в БАД из экстрактов лиственницы
К флаванонолам относятся дигидрокверцетин, дигидрокемпферол, эриодиктиол.
Приборы и реактивы
Жидкостной хроматограф с насосом высокого давления с подачей растворителя от 0,1 до 5,0 см3/мин., обеспечивающий работу в режиме градиентного элюирования, оборудованный спектрофотометрическим детектором с переменной длиной волны и системой для сбора и обработки хроматографических данных Мультихром, версия 1,5х (Амперсенд, Россия).
Колонка и предколонка хроматографические с силикагелем, химически связанным с октадецилсиланом (силикагель С18) с размером частиц 5 мкм; длина колонки - 15 см, предколонки - 4,5 см, внутренний диаметр колонок - 0,46 см;
Микрошприцы МШ-10 и МШ-25 для жидкостной хроматографии.
ГСО дигидрокверцетина ВФС 42-2399-94.
Колбы мерные наливные 2-50-2, 2-100-2, 2-250-2, 1-1000-2 по ГОСТ 1770.
Пипетки 4-1-2 или 5-1-2, 4-2-10 или 5-2-10, 4-2-25 или 5-2-25 по ГОСТ 29227.
Ацетонитрил, ч., ТУ 6-09-3534-74.
Спирт этиловый ректификованный.
Кислота уксусная ледяная, х.ч., ГОСТ 6709-72, раствор 2 %.
Кислота соляная, х.ч., ГОСТ 3118.
Капроновый фильтр марки 0,45 мкм RC.
Фумажные фильтры обеззоленные марки ФОМ.
Цинк гранулированный, ч.
Приготовление стандартного раствора
0,05±0,0001 г дигидрокверцетина, предварительно высушенного при 105 °С в течение 1 ч, помещают в мерную колбу вместимостью 100 см3, заполненную до половины ацетонитрилом, перемешивают до полного растворения образца, доводят до метки ацетонитрилом и тщательно перемешивают. 1 см3 полученного раствора концентрацией 0,5 мг/см3 переносят в мерную колбу вместимостью 25 см3, доводят ацетонитрилом до метки и тщательно перемешивают. Концентрация рабочего стандартного раствора дигидрокверцетина 0,02 мг/см3. В хроматограф вводят 10 мкл.
Подготовка образца
1 г измельченных таблеток или содержимого капсул помещают в мерную колбу вместимостью 250 см3, добавляют 50 см3 подвижной фазы ацетонитрил - 2 %-ная уксусная кислота (30:70), встряхивают 30 мин, доводят подвижной фазой до метки и тщательно перемешивают. Полученный раствор фильтруют через капроновый фильтр или центрифугируют при 3000 об/мин 5 мин. Раствор используют для проведения качественной реакции на флаванонолы (дигидрокверцетин) или анализируют с помощью ВЭЖХ.
Проведение качественной реакции
1 г измельченного образца встряхивают с 50 см3 спирта 96 %-ного, раствор фильтруют через бумажный фильтр «синяя лента», к 1 см3 фильтрата добавляют 0,5 см3 концентрированной соляной кислоты и 0,05 г цинка гранулированного. О наличии в растворе флаванонолов свидетельствует малиновое окрашивание.
Проведение анализа ВЭЖХ
Условия хроматографического анализа: колонка Hypersil ODS, размер колонки 250´4,6 мм, размер частиц 5 мкм, подвижная фаза ацетонитрил - 2 %-ная уксусная кислота (30:70), скорость подвижной фазы 1,0 см /мин., объем петли инжектора 20 мм3. УФ детектор, длина волны 287 нм. Времена удерживания флаванонолов: дигидрокверцетин - 5,0±0,1 мин, дигидрокемпферол - 6,5±0,1 мин, эриодиктиол - 7,5±0,1 мин.
При количественном анализе используют метод абсолютной калибровки. Относительное стандартное отклонение, вычисленное по площадям пиков не менее двух параллельных измерений дигидрокверцетина не должно превышать 0,4 %.
11.4. Определение изофлавонов в БАД
Принцип метода
Метод определения массовой концентрации изофлавонов в соясодержащих продуктах и БАД основан на применении высокоэффективной жидкостной хроматографы (ВЭЖХ). К изофлавонам относятся даидзеин, генистеин и глицитеин и их гликозиды даидзин, генистин, глицитин, структура которых указана на рис. 13.
Средства измерений, материалы и реактивы
Жидкостной хроматограф с насосом высокого давления с подачей растворителя от 0,1 до 5,0 см3/мин., обеспечивающий работу в режиме градиентного элюирования, оборудованный спектрофотометрическим детектором с переменной длиной волны и системой для сбора и обработки хроматографических данных Мультихром, версия 1,5х (Амперсенд, Россия).
Колонка и предколонка хроматографические с силикагелем, химически связанным с октадецилсиланом (силикагель С18) с размером частиц 5 мкм; длина колонки - 15 см, предколонки - 4,5 см, внутренний диаметр колонок - 0,46 см.
Микрошприцы МШ-10 и МШ-25 для жидкостной хроматографии.
Аппарат для встряхивания проб типа АВУ-6С, ТУ 64-1-2451-78.
Ультразвуковая баня.
Колбы мерные наливные 2-50-2, 2-100-2, 2-250-2, 1-1000-2 по ГОСТ 1770.
Пипетки 4-1-2 или 5-1-2, 4-2-10 или 5-2-10, 4-2-25 или 5-2-25 по ГОСТ 29227.
Ацетонитрил, ч., ТУ 6-09-3534-74.
Метанол - яд для жидкостной хроматографии, ос.ч. по ТУ 6-09-14-2192-85.
Бумага фильтровальная «красная лента» и «синяя лента» по ТУ 6-09-1678-95.
Фильтры капроновые марки 0,45 мкм RC или целлюлозные.
Баллон с сжатым азотом марки ПНГ.
Вода деионизированная.
Кислота о-фосфорная, ч., ГОСТ 6552.
Подготовка к проведению измерений
Приготовление рабочих растворов изофлавонов
Навеску изофлавона (генистеин) массой 10,0±0,1 мг помещают в мерную колбу вместимостью 25 см3, до половины заполненную 70 %-ным метанолом, перемешивают до полного растворения вещества, доводят 70 %-ным метанолом до метки и тщательно перемешивают. Получают раствор с массовой концентрацией генистеина 0,4 мг/см3.
Подготовка образца
Содержимое капсул или растертых таблеток БАД или 1,0 г измельченного образца помещают в круглодонную колбу вместимостью 100 см3, прибавляют около 50 см3 70 %-ного метанола, экстрагируют с помощью ультразвука в течение 10 минут и встряхивают 20 минут на аппарате для встряхивания. Экстракт охлаждают до комнатной температуры, надосадочную жидкость фильтруют через целлюлозный или капроновый фильтр с размером пор до 0,45 мкм в мерную колбу вместимостью 100 см3. Объем экстракта доводят 70 %-ным метанолом до метки и тщательно перемешивают.
Проведение анализа
Условия ВЭЖХ. Колонка с сорбентом Kromasil 100 С18 или аналогичная, размер частиц 5 мкм. Подвижная фаза: градиент 0,1 % фосфорной кислоты - ацетонитрил от (90:10) до (65:35) за 40 мин., скорость потока 0,8 см3/мин, температура колонки 25 °С. Детектор спектрофотометрический, длина волны УФ 255 нм, шкала 0,01 AUFS. Параметры разделения изофлавонов: даидзин 9,5±0,5, глицитин 10,5±0,5, генистин 14,5±0,5, даидзеин 23,5±0,5 мин., глицитеин 24,5±0,5 и генистеин 31,5±0,5 мин.
Расчет концентраций изофлавонов
Массовую концентрацию каждого изофлавона относительно стандарта генистеина рассчитывают по формуле:
![]() %,
где
%,
где
![]() - площадь пика исследуемого компонента;
- площадь пика исследуемого компонента;
![]() - площадь пика стандарта генистеина;
- площадь пика стандарта генистеина;
V1 - объем анализируемого раствора пробы, мм3;
V2 - объем экстракта, введенного в хроматограф, мм3;
![]() - масса стандарта генистеина, введенная в хроматограф,
мг;
- масса стандарта генистеина, введенная в хроматограф,
мг;
![]() - навеска образца, взятая для анализа, мг,
- навеска образца, взятая для анализа, мг,
![]() -
коэффициент пересчета содержания индивидуального изофлавона относительно
генистеина - согласно нижеприведенной таблице:
-
коэффициент пересчета содержания индивидуального изофлавона относительно
генистеина - согласно нижеприведенной таблице:
|
Изофлавон |
К |
|
Генистеин |
1,000 |
|
Генистин |
1,458 |
|
Даидзеин |
1,203 |
|
Даидзин |
1,931 |
|
Глицитеин |
1,630 |
|
Глицитин |
2,118 |
Вычисления проводят до второго десятичного знака. За результат принимают среднее арифметическое результатов двух параллельных измерений и выражают целым числом с одним десятичным знаком.
Относительное допустимое расхождение между результатами двух параллельных определений при доверительной вероятности Р = 0,95 не должно превышать 10 %.
11.5. Определение гиперозида и рутина в БАД, содержащих боярышник (Crataegus Monoguna)
Приборы и реактивы
Высокоэффективный жидкостной хроматограф с колонкой ODS (Nucleosil) 250´4,6 мм, предколонкой ODS 20´4,6 мм, размер частиц 5 мкм, объем петли инжектора 20 мкл, фотометрический детектор, длина волны 590 нм.
Пластины для ТСХ «Silufol».
Облучатель ультрафиолетовый.
Спектрофотометр.
Реактив Neu: а) смесь аминоэтанол дифенилбората, 1 %-ный раствор в метаноле и полиэтиленгликоль 400R, 5 %-ный раствор в метаноле; б) алюминий хлористый 5 %-ный раствор в спирте.
Кислота уксусная ледяная.
Кислота муравьиная безводная.
Этилацетат.
Стандартные растворы
Гиперозид, раствор в метаноле концентрацией 0,5 мг/см3.
Гиперозид, раствор в 96 %-ном этиловом спирте концентрацией 1 мг/см3.
Витексин-2-рамнозид, раствор в метаноле концентрацией 0,02 мг/см3.
Рутин, раствор 0,05 %-ный в метаноле.
Подготовка образца
1) 1,0 г измельченного продукта помещают в плоскодонную колбу, добавляют 10 см3 метанола, смесь перемешивают в течение 15 мин. Раствор фильтруют.
2) 0,5 г измельченного продукта кипятят в течение 15 мин в 5 см3 95 %-ного этилового спирта, экстракт фильтруют (для качественного анализа).
3) 2 г измельченного продукта нагревают с обратным холодильником на кипящей водяной бане в течение 1 ч в 100 см3 95 %-ного этилового спирта, после охлаждения доводят объем до первоначального, экстракт фильтруют, 50 см3 фильтрата упаривают до 2 - 3 см3, переносят в мерную колбу на 10 см3 доводят объем до 10 см3 96 %-ным этиловым спиртом, перемешивают и дают осесть образовавшемуся аморфному осадку.
4) На стартовую линию пластины Silufol (20´20 см), отстоящую от края пластины на 20 - 25 мм микрошприцем или стеклянным капилляром наносят в виде полосы 80 мкл экстракта и 80 мкл контрольного раствора ГСО гиперозида. Пластинку помещают в хроматографическую камеру и проводят разделение в системе хлороформ - метиловый спирт (8:2). Когда фронт растворителя дойдет до края пластинки, ее высушивают в течение 2 мин и вторично хроматографируют в той же системе. Высушивают и просматривают в УФ-свете при 360 нм. Отмечают пятна гиперозида в образце и стандарте. Элюируют вещество со слоя сорбента 10 см3 смеси диоксан - вода (1:1) (раствор А).
Идентификация тестового соединения
На стартовую линию пластины Silufol (20´20 см), отстоящую от края пластины на 20 - 25 мм с помощью микрошприца или стеклянного капилляра наносят в виде полосы 20´3 мм 20 мкл анализируемого экстракта и 5 мкл контрольного раствора. Пластинку помещают в хроматографическую камеру и элюируют в системе ледяная уксусная кислота - муравьиная кислота безводная - метилэтилкетон - этилацетат (10:10:30:50). Дают фронту растворителя подняться на 10 - 15 см, затем пластину высушивают, после испарения растворителя помещают на 2 мин. под струю горячего воздуха и опрыскивают реактивом Neu. В длинноволновом УФ-свете (длина волны 365 нм) гиперозид обнаруживают по полосе с желтой флуоресценцией и Rf 0,5, 2,2-рамнозил витексина - по зеленоватой и Rf 0,25, хлорогеновую кислоту - по белой и Rf 0,4 и изохлорогеновую кислоту - по голубой флуоресценции и Rf 0,7.
В системе хлороформ: метанол (8:2) на уровне гиперозида в УФ-свете 360 нм наблюдают пятно темно-коричневого цвета. При обработке пластинки 5 % спиртовым раствором AlСl3 и последующим нагреванием при 100 - 105 °С - 2 - 3 мин, пятно приобретает ярко-желтую окраску при дневном свете и желто-зеленую флуоресценцию в УФ-свете 360 нм.
Количественное определение тестовых соединений
Содержание гиперозида
Измеряют оптическую плотность исследуемого раствора А при 365 нм, против раствора сравнения - контрольный опыт (экстракт полосы сорбента). Калибровку провопят по раствору стандарта ГСО гиперозида.
Содержание витексин-2-рамнозида и гиперозида
0,5 г измельченного продукта помещают в плоскодонную колбу, соединенную с обратным холодильником, добавляют 50 см метанола, смесь нагревают на водяной бане при перемешивании в течение 30 мин. Раствор охлаждают, фильтруют, растворитель упаривают на ротационном испарителе до небольшого объема. Остаток растворяют в 25 см3 метанола, а затем разбавляют подвижной фазой в соотношении 1:5 и анализируют с помощью ВЭЖХ.
Условия хроматографического разделения: колонка ODS (Nucleosil) 250´4,6 мм, предколонка ODS 20´4,6 мм, размер частиц 5 мкм, объем петли инжектора 20 мкл УФ-детектор, длина волны 590 нм, подвижная фаза:
А: вода - тетрагидрофуран - этанол - фосфорная кислота (90:10:5:1);
В: вода - тетрагидрофуран - пропанол-2 - фосфорная кислота (20:80:5:1).
Градиент элюирования: А®В 0:100 за 45 мин, скорость потока 1 см3/мин.
Ориентировочные времена удерживания:
витексин-2-рамнозида - 22,59 мин, гиперозида - 31,90 мин.
Записывают хроматограммы стандартного образца и экстракта. Отмечают пик, совпадающий по времени удерживания с соответствующим стандартом и по площадям пиков засчитывают содержание в продукте.
11.6. Определение флавонолгликозидов в БАД на основе экстракта Ginkgo biloba
Согласно фармакопее США экстракт листьев Ginkgo biloba должен содержать не менее, чем 22,0 % и не более, чем 27,0 % флавоноловых гликозидов (рис. 14). В стандартизованных экстрактах Ginkgo biloba, содержание кверцетина, кемпферола и изорамнетина колеблется около 9,5 %, 10,5 % и 2,0 %, соответственно. Такое качественное и количественное соотношение флавонолгликозидов уникально для высших растений и может служить специфическим индикаторным показателем качества препаратов гинкго.
В качестве метода анализа используется высокоэффективная обращенно-фазная жидкостная хроматография.
Подготовка проб для анализа
Содержимое капсул или растертые таблетки, содержащие около 0,3 г сухого экстракта или 1,0 г порошка листьев гинкго помещают в круглодонную колбу объемом 250 см3, прибавляют 80 см3 смеси метанол - вода - соляная кислота (60:20:8). Экстракцию проводят на кипящей водяной бане с обратным холодильником в течение 1 - 1,5 ч. Экстракт охлаждают до комнатной температуры, надосадочную жидкость фильтруют через целлюлозный или тефлоновый фильтр с размером пор до 0,45 мкм в мерную колбу объемом 100 см3. Объем экстракта доводят дистиллированной водой до 100 см3.
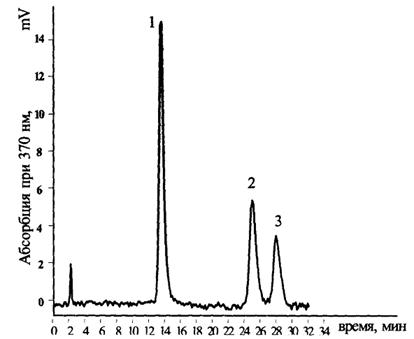
Рис. 16. Хроматограмма модельной смеси:
кверцетин (1), кемпферол (2), изорамнетин (3).
Приготовление рабочих стандартных растворов
Точные навески 1 мг стандартных образцов кверцетина, кемпферола и изорамнетина переносят в мерные колбы, встряхивают с метанолом до полного растворения, доводят объем растворителя до метки и тщательно перемешивают.
Проведение анализа
Колонка: Kromosil C18, 5 мкм, 250 мм ´ 4,6 мм ID;
Подвижная фаза: метанол - ортофосфорная кислота, рН 2,6 (53:47), скорость потока 1,2 см3/мин.
Детектирование: UV, 370 нм.
Объем вводимой пробы 5 - 20 мкл.
Параметры разделения кверцетина, кемпферола и изорамнетина в табл. 37.
Таблица 37
|
tR, мин |
К - коэффициент емкости колонки |
|
|
Кверцетин |
13,3 |
5,4 |
|
Кемпферол |
24,0 |
10,6 |
|
Изорамнетин |
27,9 |
12,5 |
Таблица 38
Метрологические характеристики метода определения флавонолгликозидов
|
Статистический параметр |
Кверцетин |
|
Предел обнаружения, % |
0,01 |
|
Интервал определяемых концентраций, мг/кг |
0,01 - 15 |
|
Среднее значение открываемости (показатель правильности), % |
90 - 95 |
|
Максимально допустимое расхождение между результатами двух параллельных внутрилабораторных определений (сходимость), при р = 0,95, % |
7,0 |
|
Максимально допустимое расхождение между результатами двух параллельных межлабораторных определений, (воспроизводимость) при р = 0,95, % |
10,0 |
Для определения содержания флавонолгликозидов найденные концентрации кверцетина умножают на коэффициент 2,5, кемпферола - на 2,05, изорамнетина - на 2,4.
11.7. Определение флавоноидов в БАД, содержащих солодку (Radix Glicyrrisae)
Солодка содержит тритерпеноидные сапонины (4 - 24 %), главным образом, глицирризин, смесь натриевой и кальциевой солей глицирризиновой кислоты; флавоноиды (1 %), в основном флаваноны, ликвиритин и ликвиритигенин, халконы изоликвиритин, изоликвиритигенин и изофлавоноиды (формононетин).
Анализ основан на определении глицерризиновой кислоты и тестовых соединений с помощью ТСХ и ВЭЖХ.
Приборы и реактивы
Спектрофотометр СФ-26, СФ-46 или подобный (Shimadzu UV-160A, Япония), позволяющий проводить измерения при длинах волн 190 - 900 нм, с допустимой абсолютной погрешностью измерений коэффициента пропускания не более 1 %;
Пластины «Silufol».
Жидкостной хроматограф с насосом высокого давления с подачей растворителя от 0,1 до 5,0 см3/мин., обеспечивающий работу в режиме градиентного элюирования, оборудованный спектрофотометрическим детектором с переменной длиной волны и системой для сбора и обработки хроматографических данных Мультихром, версия 1,5х (Амперсенд, Россия).
Колонка и предколонка хроматографические с силикагелем, химически связанным с октадецилсиланом (силикагель С18) с размером частиц 5 мкм; длина колонки - 15 см, предколонки - 4,5 см, внутренний диаметр колонок - 0,46 см.
Микрошприцы МШ-10 и МШ-25 для жидкостной хроматографии.
Спирт этиловый ректификованный.
Метанол - яд для жидкостной хроматографии, ос.ч. по ТУ 6-09-14-2192-85.
Бумага фильтровальная «красная лента» и «синяя лента» по ТУ 6-09-1678-95
Ликуразид, раствор в 50 %-ном этиловом спирте концентрацией 1 мг/см3.
Глицирризиновая кислота, аммониевая или натриевая соль: раствор в 50 %-ном этиловом спирте концентрацией 1 мг/см3.
Подготовка образца
1 г измельченного продукта помещают в плоскодонную колбу, добавляют 50 см3 50 %-ного этилового спирта, смесь нагревают на водяной бане при 55 - 60 °С в течение 30 мин. Экстракцию повторяют пятикратно. Спиртовые экстракты, охлаждают, фильтруют, доводят объем до 250 см3 50 %-ным этиловым спиртом.
Идентификация тестовых соединений ТСХ
На стартовую линию пластины Silufol-УФ 254 (20´20 мм), отстоящую от края пластины на 20 - 25 мм с помощью микрошприца или стеклянного капилляра наносят в виде полосы 20´3 мм 10 мкл экстракта и по 5 мкл растворов стандартов: ГСО ликуразида, глицирризиновой кислоты. Пластинку помещают в хроматографическую камеу и элюируют в смеси хлороформ: метанол: вода, взятых в объемном соотношении 61:32:7. Дают фронту растворителя подняться на 10 см, затем после высушивания пластинку рассматривают в видимом и УФ свете при длинах волн 254 и 360 нм: наблюдают желтые (видимый свет) или темно-фиолетовые (УФ свет) пятна с Rf 0,9 - изоликвиритигенин, Rf 0,5 - изоликвиритин, Rf 0,4 - ликуразид. Только в УФ-свете проявляются синим цветом пятна: глицирризиновой кислоты - Rf 0,1, ликвиритигенин Rf 0,8, ононин Rf 0,6, ликвиритин Rf 0,5. При опрыскивании пластины диазореагентом все вещества, кроме ононина, проявляются в виде желтых или оранжевых пятен.
Количественное определение ВЭЖХ.
Условия хроматографического разделения: колонка Zorbax ODS, 5 мкм, 250´4,6 мм, предколонка - ODS, 5 мкм, 20´4,6 мм, объем петли инжектора 20 мкл. Подвижная фаза: градиент метанол - 1,5 % уксусная кислота от (20:80) до (40:60) 60 мин, скорость потока 1 см3/мин, температура колонки 45 °С. УФ-детектор, длины волн: 275 нм для обнаружения ликвиритина и 375 нм для обнаружения ликуразида. Время выхода 10 и 25 мин. соответственно.
11.8. Определение флавоновых гликозидов в БАД, содержащих страстоцвет (Passiflora incarnata L)
Приборы и реактивы
Пластины для ТСХ с силикагелем («Silufol», «Sorbfil»).
Реактив Neu: а) смесь 1 %-ного раствора дифенилбората аминоэтанола в метаноле и 5 %-ного раствора полиэтиленгликоля - 400R (PEG-400R) в метаноле; б) 2 %-ный раствор хлорида алюминия в метаноле.
Стандартные растворы в метаноле витексина, изовитексина, сапонарина концентрацией 0,5 - 1,0 мг/см3.
Подготовка образца
К 1 г порошка БАД добавляют 10 см3 метанола, нагревают с обратным холодильником и при перемешивании на водяной бане при 60 °С в течение 10 мин. Экстракт фильтруют через сульфат натрия и упаривают до 1 см3.
К 20 г порошка БАД добавляют 200 см3 этилового спирта, кипятят с обратным холодильником на водяной бане в течение 1 ч, фильтруют и упаривают досуха. Остаток растворяют при перемешивании в 0,5 %-ной серной кислоте в течение 15 мин, раствор фильтруют, фильтрат подщелачивают 20 %-ным КОН до рН 9 и экстрагируют хлористым метиленом (3´50 см3). Экстракт обезвоживают сульфатом натрия и упаривают досуха. Остаток растворяют в 1 см3 метанола.
Идентификация тестового соединения
На стартовую линию пластины Silufol (20´20 см), отстоящую от края на 20 - 25 мм с помощью микрошприца или стеклянного капилляра наносят в виде полосы 20´3 мм по 20 мкл экстракта и эталонного раствора витексина, изовитексина или сапонарина. Пластинку помещают в хроматографическую камеру и элюируют в системе растворителей безводная муравьиная кислота - вода - этилацетат - метилэтилкетон (10:20:50:30). Дают фронту растворителя подняться на 12 см, пластинку высушивают и опрыскивают проявляющим реагентом. На хроматограмме в УФ-свете (365 нм) флавоновые гликозиды проявляются в виде зеленых пятен изовитексин с Rf 0,5, витексин - с Rf 0,65, сапонарин - с Rf 0,15.
Количественное определение суммы флавоновых гликозидов
Количественное определение суммы флавоновых гликозидов проводят с помощью метода спектрофотометрии, согласно разделу 10.
12. Определение содержания гинзенозидов в БАД, содержащих женьшень
В настоящее время корни культивируемого и дикорастущего многолетнего травянистого растения женьшеня (Рапах ginseng C.A. Mey, сем. аралиевых - Araliaceae) и его экстракты используются в качестве добавок при изготовлении ряда пищевых продуктов, а также входят в состав биологически активных добавок к пище. Его биологическая активность связывается, в основном, с соединениями группы тритерпеновых гликозидов (гинзенозидов). Основными гинзенозидами женьшеня являются Re, Rg1, Rg1, Rf, Rg2, Rb1, Rс, Rb2, Rd, остальные присутствуют в минорных концентрациях. Для стандартизации корня женьшеня и его препаратов обычно определяют содержание шести гинзенозидов - Re, Rg1, Rb1, Rс, Rb2, Rd. В настоящее время стандартные образцы существуют только для Re, Rb1 и Re (рис. 17).
В качестве метода анализа используется высокоэффективная обращенно-фазная жидкостная хроматография. Предел обнаружения содержания гинзенозидов Rb1, Re, Re составляет 0,001 мг/см3. Отклик детектора линеен в диапазоне концентраций гинзенозидов от 0,001 до 1,00 мг/см3.
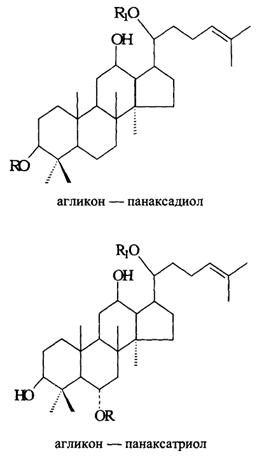
|
Название |
R |
R1 |
|
Rb1 |
D-Glc-b-(l®2)-D-Glc |
D-Glc-b (l®6)-D-Glc |
|
Rb2 |
D-Glc-b (l®2)-D-Glc |
L-Аrа (пираноза) (1®6)-D-Glc |
|
Rс |
D-Glc-b (l®2)-D-Glc |
L-Аrа (фураноза) (1®6)-D-Glc |
|
Rd |
D-Glc-b (l®2)-D-Glc |
D-Glc |
|
Название |
R |
R1 |
|
Re |
L-Rha (l®2)-D-Glc |
D-Glc |
|
Rf |
D-Glc-b (l®2)-D-Glc |
D-Glc |
|
Rg1 |
D-Glc |
D-Glc |
|
Rg2 |
L-Rha (l®2)-D-Glc |
H |
Рис. 17.
Подготовка проб для анализа
Напитки и биологически активные добавки к пище в жидком виде
10 см3 напитка с женьшенем наносят на патрон Диапак С18, предварительно активированный 10 см3 метилового спирта и 20 см3 дистиллированной воды. Патрон промывают 20 см3 дистиллированной воды. Гинзенозиды смывают 10 см3 метилового спирта в круглодонную колбу вместимостью 25 см3. Раствор отгоняют досуха на роторном испарителе при температуре не более 50 °С. Сухой остаток в колбе растворяют в 1 см3 метилового спирта. 200 мкл полученного раствора помещают поверх мембранного фильтра в патрон набора для микрофильтрации образцов на 1,0 см3 (НПФ «БИОХРОМ», Москва, Россия) и центрифугируют на микроцентрифуге-вортекс «MINIGEN» (фирма «Диа-М», Москва, Россия) или Опн-8 при 3000 об/мин в течение 10 мин.
Биологически активные добавки к пище в сухом виде
Определение содержания суммы гинзенозидов Rb1, Re, Re
0,005 г (точная навеска) препарата помещают поверх мембранного фильтра в патрон набора для микрофильтрации образцов на 1,0 см3. Добавляют 400 мкл гексана и центрифугируют на микроцентрифуге-вортекс «MINIGEN» при 3000 об/мин в течение 3 мин. Промывку гексаном повторяют трижды. Затем патрон для образцов сушат под вакуумом до удаления запаха гексана. Фильтр с образцом количественно переносят в герметически закрывающуюся пробирку вместимостью 5 см3 и добавляют 2 см3 дистиллированной воды. Содержимое пробирки перемешивают на микроцентрифуге-вортекс «MINIGEN» или аналогичной в течение 1 мин. 1 см3 раствора помещают в микропробирку из полипропилена объемом 1,5 см3 и центрифугируют на микроцентрифуге-вортекс «MINIGEN» или Опн-8 при 3 000 об/мин в течение 10 мин.
Мед
10±0,01 г меда с женьшенем помещают в коническую колбу вместимостью 100 см3 и растворяют в 50 см3 воды дистиллированной. 40 см3 полученного раствора наносят на патрон Диапак С18, предварительно активированный 10 см3 метилового спирта и 20 см3 дистиллированной воды. Далее проводят те же операции, которые были описаны выше для напитков и биологически активных добавок к пище в жидком виде.
Приготовление рабочих стандартных растворов
Для приготовления раствора РСО гинзенозида Rb1 (Rс или Re) в фирменный флакон с РСО соответствующего гинзенозида (Sigma, кат. № G-0777, G-0902 и G-1027) прибавляют 5 см3 метилового спирта и перемешивают до растворения. Срок годности раствора РСО при хранении его при температуре не выше 4 °С - 1 мес.
Проведение анализа
Колонка: Кромасил 100 С18 (Kromasil), 7 мкм, 150´4 мм ID.
Элюент: ацетонитрил - 0,05 % ортофосфорная кислота (30:70), 1,0 см3/мин.
Детектирование: UV, 203 нм.
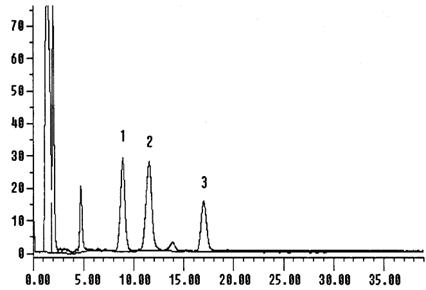
Рис. 18. Хроматограмма модельной смеси гинзенозидов Rb1 (I), Rс (2), Re (3).
Таблица 39
Метрологические характеристики методики определения гинзенозидов Rb1, Rс, Re в корнях женьшеня
|
Статистический параметр |
Rb1, Rс, Re |
|
Предел обнаружения, мг/мл |
0,001 |
|
Интервал определяемых концентраций, мг/мл |
0,001 - 1,00 |
|
Среднее значение открываемости (показатель правильности), % |
95,5 |
|
Максимально допустимое расхождение между результатами двух параллельных внутрилабораторных определений (сходимость), при р = 0,95, % |
7,0 |
|
Максимально допустимое расхождение между результатами двух параллельных межлабораторных определений, (воспроизводимость) при р = 0,95, % |
10,0 |
13. Определение содержания схизандрина в БАД, содержащих лимонник китайский (Schizandra Chinensis (Turcz.) Baill)
Метод основан на определении схизандрина с помощью метода ВЭЖХ.
Температура плавления схизандрина 133 °С; [a]![]() + 78 °С (с 1,0; хлороформ); склонен удерживать кристаллизационную вод даже при
нагревании в вакууме. В УФ-спектре максимумы длин волн в диапазонах 221 - 224
нм и 249 - 251 нм.
+ 78 °С (с 1,0; хлороформ); склонен удерживать кристаллизационную вод даже при
нагревании в вакууме. В УФ-спектре максимумы длин волн в диапазонах 221 - 224
нм и 249 - 251 нм.
Подготовка проб для анализа
Напитки и биологически активные добавки к пище в жидком виде, содержащие лимонник, его экстракты, настои и настойки
1 см3 пробы переносят в пробирку для микропроб однократного применения объемом 1,5 см3 и центрифугируют на микроцентрифуге-вортексе «MINIGEN» (фирма «Диа-М», Москва, Россия) или на центрифуге медицинской лабораторной ОПн-8 при 3000 об/мин в течение 10 мин. Если при анализе пик схизандрина не обнаружен, то раствор предварительно центрифугируют и супернатант упаривают досуха на роторном испарителе. Остаток растворяют в 1 - 3 см3 30 % этилового спирта.
Биологически активные добавки к пище в сухом виде, содержащие лимонник, его экстракты, настои и настойки
Навеску 0,5 г ±0,001 биологически активной добавки к пище помещают в коническую колбу вместимостью 25 см3 и приливают 5 см3 30 % этилового спирта. Содержимое колбы перемешивают в течение 5 мин. 1 см3 раствора переносят в пробирку для микропроб однократного применения объемом 1,5 см3 и центрифугируют при 3000 об/мин в течение 10 мин.
Мед, содержащий лимонник, его экстракты, настои и настойки
Навеску 10±0,01 г меда растворяют в 25 см3 дистиллированной воды при нагревании. Концентрирующий патрон Диапак С16 активируют 10 см3 спирта и отмывают 10 см3 воды. 20 см3 водного раствора образца наносят на патрон концентрирующий Диапак С16, порциями по 10 см3, после каждой порции патрон промывают 20 см3 воды. Вещества элюируют с патрона 10 см3 спирта и помещают в круглодонную колбу вместимостью 25 см3. Раствор упаривают досуха на роторном испарителе. В колбу приливают 2 см3 спирта и растворяют сухой остаток. 1 см3 раствора помещают в пробирку для микропроб однократного применения объемом 1,5 см3 и центрифугируют при 3000 об/мин в течение 10 мин.
Приготовление рабочих стандартных растворов
5±0,1 мг стандартного образца схизандрина переносят в мерную колбу на 10 см3, растворяют в небольшом количестве метанола и затем доводят до метки метанолом. Получают раствор РСО с концентрацией 0,5 мг/см3. Раствор РСО хранят в темном месте в холодильнике в посуде с пришлифованной пробкой не более 3 мес. В хроматограф вводят 5 - 10 мкл раствора образца.
Проведение анализа
Предколонка: мини-картридж Элсикарт, 8´4 мм, Диасорб 130 С16Т;
Колонка: Диасорб 130 С16Т, 11 мкм, 250´4 мм ID;
Подвижная фаза - ацетонитрил - вода (50:50), 1,0 см3/мин;
Детектирование: UV, 216 нм.
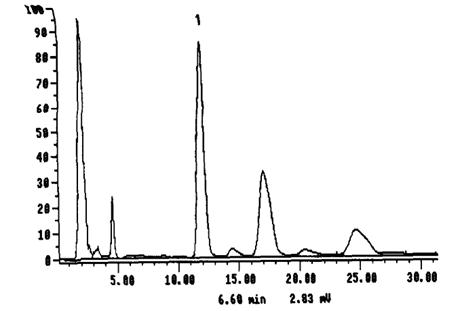
Рис. 19. Хроматограмма экстракта лимонника (пик 1 - схизандрин).
Время удерживания схизандрина составляет около 12 мин.
Таблица 40
Метрологические характеристики методики определения схизандрина в плодах, семенах и лианах лимонника
|
Статистический параметр |
Схизандрин |
|
Предел обнаружения, мг/мл |
0,001 |
|
Интервал определяемых концентраций, мг/мл |
0,001 - 1,00 |
|
Среднее значение открываемости (показатель правильности), % |
95,5 |
|
Максимально допустимое расхождение между результатами двух параллельных внутрилабораторных определений (сходимость), при р = 0,95 % |
7,0 |
|
Максимально допустимое расхождение между результатами двух параллельных межлабораторных определений (воспроизводимость) при р = 0,95, % |
10,0 |
14. Определение содержания элеутерозида В (сирингина) в БАД, содержащих элеутерококк колючий
Компонентами элеутерококка являются стероиды (даукостерин), фенилпропаноиды с замещенными фенольными группами (сирингин), лигнаны (элеутерозид D), оксикумарины (7-О-b-D-глюкозилизофраксидин), алканы (этил-a-D-галактозид или элеутерозид С), а также негликозилированные соединения различных классов: фенилпропаноиды со свободными фенольными гидроксилами (хлорогеновая кислота, конифериловый альдегид, этиловый эфир кофейной кислоты, синаповый спирт), лигнаны (сирингарезинол, сезамин), тритерпеноиды (олеаноловая кислота), кумарин (изофраксидин), ситостерины (b-ситостерин, кампестерин), полисахариды и др.
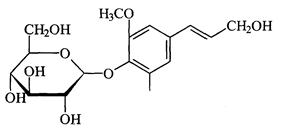
Рис. 20. Сирингин (элеутерозид В) С17Н24О9,М.м. 372,00 Т.пл. 192 - 194 °С
Сирингин мало и медленно растворим в воде и в 95 %-ном спирте.
В качестве метода анализа используют высокоэффективную обращенно-фазную жидкостную хроматографию. Предел обнаружения элеутерозида В составляет 0,001 мг/см3. Диапазон определяемых концентраций - от 0,001 до 1,00 мг/см3.
Приборы и реактивы
Облучатель ультрафиолетовый.
Пластины для ТСХ «Сорбфил ПТСХ-П-А» 10´15 см.
Пластины Silufol.
ВЭЖХ с УФ-детектором.
Приготовление рабочего раствора сирингина (элеутерозида В)
0,01±0,0005 г сирингина помещают в мерную колбу вместимостью 50 см3, прибавляют 40 см3 60 % этилового спирта, перемешивают до растворения и доводят объем раствора до метки. Срок хранения рабочего раствора при температуре 5 - 10 °С составляет 1 мес.
Подготовка образца
К 0,2 г препарата (содержание элеутерококка 15 %) добавляют 25 см3 этилового спирта 30 % и перемешивают в течение 30 мин. Центрифугируют при 3000 об/мин 1,5 см3 экстракта в пробирке для микропроб однократного применения.
0,05 г измельченного сырья обезжиривают 3-х кратной экстракцией хлороформом или гексаном, из остатка экстрагируют элеутерозиды метанолом или 95 % этанолом (10 см3) при нагревании с обратным холодильником при 50 °С в течение 1 ч, раствор фильтруют и доводят объем до первоначального.
Идентификация тестового соединения с помощью метода ТСХ
На пластину для ТСХ полосой наносят 30 мкл экстракта, высушивают при 100 °С в течение 15 мин. Хроматографирование проводят в системе растворителей: этиловый спирт 96 % - хлороформ (7:3). Пластину сушат при 100 °С в течение 5 мин. В УФ-свете (360 нм) наблюдают бледно-голубую полоску с Rf 0,64.
Проводят хроматографирование экстракта (п. 3.2) на пластинах Silufol в системе хлороформ - метанол - вода (61:32:7). Проявление пятен производят опрыскиванием пластины 20 % серной кислотой.
Проведение ВЭЖХ
Колонка: Диасорб 130 С16Т, 11 мкм, 250´4 мм ID или аналогичная.
Подвижная фаза: ацетонитрил - вода - ледяная уксусная кислота (85:15:0,5), 1 см3/мин.
Детектирование: UV, 266 нм.
Время удерживания элеутерозида В около 12 мин.
Рис. 21. Хроматограмма экстракта элеутероккока колючего, (1) - элеутерозид В (сирингин).
Таблица 41
Метрологические характеристики методики определения элеутерозида В в напитках и БАД в сухом виде
|
Статистический параметр |
элеутерозид В (сирингин) |
|
Предел обнаружения, мг/мл |
0,001 |
|
Интервал определяемых концентраций, мг/мл |
0,001 - 1,00 |
|
Среднее значение открываемости, (правильность), % |
95,5 |
|
Максимально допустимое расхождение между результатами двух параллельных внутрилабораторных определений (сходимость), р = 0,95, % |
7,0 |
|
Максимально допустимое расхождение между результатами двух параллельных межлабораторных определений (воспроизводимость), р = 0,95, % |
10,0 |
15. Определение производных кофейной (3,4-дигидрокси-коричной) кислоты в БАД на основе экстрактов эхинацеи пурпурной
Идентификации и количественного определение производных кофейной кислоты в препаратах эхинацеи проводится с помощью метода ВЭЖХ.
Приготовление пробы
1 г измельченной травы, содержимое капсул или растертых таблеток помещают в колбу объемом 200 см3, прибавляют 100 см3 смеси метанол - вода (7:3) и ведут экстракцию при нагревании на кипящей водяной бане с обратным холодильником в течение 10 - 15 мин. Экстракт охлаждают до комнатной температуры и фильтруют через бумажный фильтр.
Приготовление рабочего стандартного образца (РСО)
В качестве внутреннего стандарта, используют 3-О-метилпроизводное кофейной кислоты - феруловую кислоту V (3-(4¢-гидрокси-3¢-метоксифенил)-проп-2-еновая кислота) - вещество близкое по хроматографической подвижности и спектральным свойствам к анализируемым производным кофейной кислоты. Для приготовления РСО с концентрацией 0,1 мг/см3, в мерную колбу на 50 см3 вносят навеску 5 мг феруловой кислоты и добавляют до метки метанола. Смесь взбалтывают до полного растворения. Концентрация РСО проверяют с помощью УФ-спектрофотометрии (длина волны максимума 234 нм (logE 4,10)).
Проведение анализа
Условия ВЭЖХ. К 1 см3 экстракта добавляют 1 см3 стандартного раствора феруловой кислоты с концентрацией 0,1 мг/см3 (100 мкг). Анализ проб проводят с помощью жидкостного хроматографа с УФ-спектрофотометрическим детектором. Колонка с силикагелем, химически связанным с октадецилсиланом «Нуклеосил С 18» (250´4,6 мм) или аналогичная. Подвижная фаза: ацетонитрил-0,05 М NaH2PO4, pH 2,6 (15:85); скорость потока 1,2 см3/мин; детектирование при l = 330 нм; объем вводимой пробы 5 - 20 мкл.
Рис. 22. Хроматограмма экстракта травы Е. purpurea с внутренним стандартом.
А - пик цикориевой кислоты; В - пик кафтаровой кислоты; С - пик хлорогеновой кислоты; D - пик феруловой кислоты (внутренний стандарт).
Содержание цикориевой, кафтаровой и хлорогеновой кислот в образце вычисляют по формуле:
![]() ,
где
,
где
С - концентрация внутреннего стандарта в анализируемой пробе, мг/мл;
S1 - площадь пика цикориевой кислоты;
S2 - площадь пика феруловой кислоты;
V - общий объем экстракта, мл;
М - масса навески, мг;
К - коэффициент, учитывающий различие в УФ-поглощении внутреннего стандарта V и анализируемых кислот I - III.
КI = 1,212 (для цикориевой кислоты)
КII = 1,548 (для кафтаровой кислоты)
КIII = 1,744 (для хлорогеновой кислоты)
Таблица 42
Метрологические характеристики методики определения 3,4-дигидроксикоричной кислоты
|
Статистический параметр |
Цикориевая кислота |
|
Предел обнаружения, % |
0,005 |
|
Интервал определяемых концентраций, % |
0,005 - 10 |
|
Среднее значение открываемости, (правильность), % |
90 - 95 |
|
Максимально допустимое расхождение между результатами двух параллельных внутрилабораторных определений (сходимость), р = 0,95, % |
8,0 |
|
Максимально допустимое расхождение между результатами двух параллельных межлабораторных определений (воспроизводимость), р = 0,95, % |
10,0 |
16. Определение берберина и иохимбина в БАД
Описание берберина
Светло-желтые кристаллы. Растворимость: растворим в воде; легко растворим в спирте; очень легко растворим в горячем спирте и воде; растворим в бензоле, хлороформе, эфире. Алкалоид встречается в корневищах канадского желтокорня (Hydrastis canadensis), а также в растениях сем. Лютиковых (Ranunculaceae), барбарисовых (Веrberidaceae), лунносемянниковых (Menispermaceae), рутовых (Rutaceae), и др.
Описание берберина гидрохлорида
Из H2О кристаллизуется в виде игл. Растворимость: практически не растворим в спирте. UV спектр - 228 нм (Е1 % 814), 263 нм (Е1 % 794), 345 нм (Е1 % 722), 421 нм (Е1 % 160) (данные по стандарту на гидрохлорид берберина Национального института здравоохранения Японии Eisei Shikenjo Hokoku 1995; 113:121 - 126) (рис. 23).
Рис. 23. Берберин
Описание иохимбина C21H26O3N2, М - 354,43
Белый или беловатый кристаллический порошок, без запаха, слабо горького вкуса. Кристаллизуется в иглах из разбавленного спирта. Растворимость: трудно растворим в воде, спирте и хлороформе; растворим в эфире. Tпл 234 - 235 °С, [a]20 от 50 °С до 55 °С (С2Н5ОН); 11 °С (СНСl3); 108 °С (пиридин).
Описание иохимбина гидрохлорида
Кристаллизуется в виде орторомбических листочков или призм. Растворимость: легко растворим в воде; растворим в спирте. Tпл 288 - 290 °С (с разл.). [a]D20 100 °С (с = 1, Н2O).
Рис. 24. Иохимбин
Подготовка проб для хроматографического анализа
Методика включает предварительную экстракцию алкалоидов из препаратов БАД смесями растворителей, близкими по составу к подвижной фазе при ВЭЖХ. Полноту экстракции алкалоидов оценивали по методике Британской фармакопеи (British Pharmacopoeia, 1990, vol., Арр. Х3 G, A112).
Для БАД, содержащих порошок коры иохимбе (корневища желтокорня)
0,5 - 1,0 г измельченного содержимого капсул или растертых таблеток помещают в коническую плоскодонную колбу вместимостью 25 см3 с притертой пробкой, прибавляют 5 - 10 см3 0,01N и нагревают на кипящей водяной бане в течение 10 - 15 мин. Водно-кислотный экстракт фильтруют через слой окиси алюминия.
Для БАД, содержащих экстракт коры иохимбе (корневища желтокорня)
0,5 - 1,0 г измельченного содержимого капсул или растертых таблеток помещают в коническую плоскодонную колбу с притертой пробкой, прибавляют 25 см3 метанола, подкисленного раствором соляной кислоты, и оставляют на 1 ч. (рассчитать количество 0,01N HCl для подкисления метанола до рН 2,9). Метанольный раствор фильтруют через слой окиси алюминия.
Приготовление стандартных растворов (РСО)
Для приготовления РСО с концентрацией 50 нг/мкл в мерную колбу на 100 см3 вносят точные навески по 0,005 г стандартных образцов иохимбина гидрохлорида (берберина гидрохлорида); добавляют по 50 см3 воды и метанола для хроматографии (ТУ 6-09-4326-76, «ХЧ»), затем взбалтывают до полного растворения. Чистоту РСО проверяют с помощью ВЭЖХ, ТСХ и УФ-спектрофотометрии. Полученные растворы РСО используют как для идентификации компонентов БАД по параметрам удерживания при ВЭЖХ, так и для количественной оценки их содержания в образцах.
Проведение анализа
ТСХ-скрининг
Для ТСХ скрининга алкалоидов применяют пластинки с силикагелем с УФ-254 нм индикатором (силуфол, Чехия).
Подвижные фазы
Берберин: этилацетат - изопропанол - уксусная кислота 20 % (4:6:1).
Иохимбин: бензол - этилацетат - изопропанол - аммиак (12:6:4:0,5).
В качестве свидетелей применяют растворы стандартных образцов (РСО).
Зоны веществ на хроматограммах детектируют с помощью реактива Драгендорфа, по флуоресценции при 366 нм и по гашению флуоресценции индикатора при 254 нм. После разделения пластинки подсушивают и детектируют в УФ-свете при длине волны 360 нм. Чувствительность метода составляет от 1,5 до 0,5 мкг в пятне каждого алкалоида.
Условия ВЭЖХ
Анализ проб проводят с помощью жидкостного хроматографа со спектрофотометрическим детектором. Колонка с силикагелем, химически связанным с октадецилсиланом «Нуклеосил С 18» (200´4,6 мм) или аналогичная.
Подвижная фаза - метанол - вода - 85 %-о-фосфорная кислота (35:65:0,2, рН 3,0).
Содержание алкалоидов определяют методом абсолютной калибровки по внешнему стандарту. В качестве стандартных образцов используют химически чистые для хроматографии - Yohimbine hydrochloride (Y3125 SIGMA), Berberine Chloride (B3251 SIGMA).
Содержание действующего вещества в 1 таблетке/капсуле в миллиграммах (X) образце вычисляют по формуле:
![]() ,
где
,
где
Нх - площадь пика соответствующего компонента в испытуемом образце;
Hst - площадь пика соответствующего компонента в растворе РСО;
mst - масса стандарта, нг;
V1 - объем испытуемого раствора, мл;
V2 - объем введенной пробы, мкл.
Пределы обнаружения алкалоидов: в таблетках БАД массой от 0,5 до 2,0 г составляют для иохимбина - 0,1 мг/табл., для берберина - 0,02 мг/табл.
17. Определение стевиозидов в БАД, содержащих стевию (Stevia rebaudiana)
Содержание стевиозида в листьях стевии 10 - 12 %, в растениях, выращенных в Средней Азии, до 18 %, в Крыму, Украине, Молдавии, Грузии 5 - 7 %. Стевиозид хорошо растворим в воде, спирте, при нагревании неустойчив.
Подготовка образца
Таблетки или содержимое капсулы перетирают в ступке до порошка и экстрагируют трижды 60 %-ным этанолом при 50 °С с обратным холодильником в течение 15 мин на водяной бане. Спиртовые экстракты объединяют, спирт отгоняют на роторном испарителе при температуре не выше 40 °С. К сухому остатку прибавляют воду, а затем хлороформ. Пробу переносят в делительную воронку и встряхивают. Сливают нижний слой и экстракцию проводят еще два раза. Хлороформ полностью отделяют (если необходимо пробу центрифугируют), к водному слою прибавляют этанол до конечной его концентрации 50 %.
Идентификация и количественное определение стевиозида
ВЭЖХ: Колонка из нержавеющей стали, длина 250 мм, диаметр 4,0 мм. Сорбент - сепарон SGX NH2, зернение 7 мкм. Перед работой новую колонку промывают 40 см3 изопропанола, затем 80 см3 деионизированной воды, после чего уравновешивают колонку подвижной фазой до стабильной нулевой линии.
Подвижная фаза: ацетонитрил - вода (80:20).
Стандартный раствор стевиозида концентрацией 200 мкг/см3 в 50 %-ном этаноле.
Детектирование: спектрофотометр, длина волны УФ 208 нм, шкала 0,02 AUFS, скорость потока 1,0 см3/мин., время удерживания стевиозида - 12,4±0,1 мин. Обработка хроматографических данных по программе МультиХром для Windows, версия 1.39.
Стандартный образец и испытываемую пробу хроматографируют не менее трех раз. Ошибка метода ±2 %.
18. Определение салидрозидов в БАД, содержащих родиолу розовую (Rhodiola rosae L)
Приборы и реактивы
Пластины для ТСХ «Silufol».
Облучатель ультрафиолетовый.
Спектрофотометр.
Раствор ацетата свинца 10 %. Раствор натрия карбоната, 2 %. Насыщенный раствор сульфата натрия.
Диазотированный сульфанил: 7 г сульфацила натрия растворяют в 50 см3 воды в мерной колбе вместимостью 100 см3, добавляют 9 см3 концентрированной соляной кислоты, доводят раствор до метки. 1 см3 полученного раствора помещают в колбу вместимостью 100 см3, ставят на лед, добавляют 50 см3 воды, 0,2 см3 10 % натрия нитрита.
Подготовка образца
1. 1,0 г измельченного образца помещают в плоскодонную колбу, добавляют 10 см3 метанола, смесь нагревают на водяной бане с обратным холодильником при температуре 65 °С и перемешивании в течение 20 мин. Раствор фильтруют.
2. 0,5 г продукта экстрагируют 10 см3 воды при нагревании с обратным холодильником на кипящей водяной бане в течение 15 мин. Раствор фильтруют или центрифугируют, осадок экстрагируют еще трижды по 10 см3 воды, объединенный экстракт фильтруют, охлаждают.
Идентификация тестовых соединений с помощью ТСХ
На стартовую линию пластины Silufol (20´20 см), отстоящую от края пластины на 20 - 25 мм с помощью микрошприца или стеклянного капилляра наносят в виде полосы 20´3 мм 10 мкл экстракта, приготовленного по п. 1. Пластинку помещают в хроматографическую камеру и элюируют в смеси хлороформа, метанола и воды, взятых в объемном соотношении 26:14:3. Дают фронту растворителя подняться на 10 см, затем после высушивания пластинку просматривают в УФ-свете (254 нм). На хроматограмме обнаруживают доминирующее пятно фиолетового цвета с Rf 0,4 - розавин. Пластинку опрыскивают 10 % водным раствором натрия карбоната, нагревают в сушильном шкафу при температуре 110 °С в течение 2 мин., опрыскивают диазотированным сульфанилом и снова нагревают 2 мин при 100 °С. На хроматограмме салидрозид наблюдают в виде полосы красноватого цвета с Rf 0,42.
Количественное определение салидрозида
К экстракту, полученному по п. 2, прибавляют 6 см3 10 % ацетата свинца, 2 см3 насыщенного раствора сульфата натрия, перемешивают, доводят объем до 50 см3, фильтруют, первые 15 см3 отбрасывают. В мерную колбу на 25 см3 переносят 5 см3 фильтрата, прибавляют 2,5 см3 натрия карбоната и 2,5 см3 диазотированного сульфанила, доводят объем до метки, перемешивают и через 5 мин измеряют оптическую плотность раствора относительно воды при 486 нм.
Содержание салидрозида, %, вычисляют по формуле:
![]() ,
где
,
где
D - оптическая плотность,
253 - удельный показатель поглощения (Е/ % см),
m - масса в г,
W - влажность, %
19. Определение дубильных веществ в БАД, содержащих черемуху (Padus Avium Mill); ольху (ALNus incana); дуб (Qercus robur); бадан (Bergenia crassifolia)
Приборы и реактивы
Раствор перманганата калия 0,02 М.
Раствор индигосульфокислоты: в мерную колбу помещают 1 г индиго кармина, 50 см3 концентрированной серной кислоты, объем доводят водой до 1 дм3.
Подготовка образца
К 2 г измельченных корней добавляют 250 см3 нагретой до кипения воды, смесь кипятят с обратным холодильником в течение 30 мин, экстракт фильтруют.
Количественное определение дубильных веществ
25 см3 раствора, полученного выше, разбавляют водой до 500 см3, добавляют 25 см3 раствора индигосульфокислоты и титруют раствором перманганата до золотисто-желтого окрашивания.
1 см3 раствора перманганата соответствует 4,157 мг таннина.
Содержание дубильных веществ по галловой кислоте
Приборы и реактивы
0,05 М раствор натрия тетрабората - буры (тетраборнокислого 10-ти водного): 19,07 г буры растворяют в 1000 см3 воды.
0,1 Н раствор соляной кислоты: 8,5 см3 соляной кислоты концентрированной (плотность 1,190) разбавляют до 1 дм3 водой.
Буферный раствор рН 9: смешивают 900 см3 0,05 М раствора буры и 100 см3 0,1 Н соляной кислоты.
Количественное определение дубильных веществ
2 - 4 см3 исследуемого раствора помещают в мерную колбу вместимостью 50 см3, добавляют 30 см 50 %-ного этилового спирта, взбалтывают до растворения, доводят до метки 50 %-ным этиловым спиртом (раствор А).
1 см3 раствора А помещают в колбу вместимостью 50 см3, доводят до метки буферным раствором (раствор В).
Через 10 мин измеряют оптическую плотность раствора В при 277 нм. В качестве раствора сравнения используют буферный раствор.
Массовую долю дубильных веществ в пересчете на галловую кислоту, % (X), определяют по формуле:
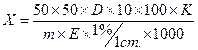 ,
где
,
где
E - удельный показатель поглощения галловой кислоты, равный 508 (оптическая плотность 1 % раствора галловой кислоты - мг/см3);
m - масса образца, г;
К - коэффициент разведения (10 = 250/25);
или по формуле
![]() ,
%, где
,
%, где
0,0197 - концентрация раствора галловой кислоты, мг/см3, оптическая плотность которого равна 1.
20. Определение производных антрахинона
20.1. В БАД, содержащих марену красильную (Rubia tinctorum L.), Марену грузинскую (Rubia iberica (FICH.EX.DС).C.COCH)
Приборы и реактивы
Спектрофотометр или ФЭК.
Щелочно-аммиачный раствор: 50 г едкого натра растворяют в 870 см3 воды, добавляют 80 см3 концентрированного раствора аммиака.
Подготовка образца
0,1 г измельченного продукта помещают в колбу на 200 см3, добавляют 7,5 см3 ледяной уксусной кислоты, 1 см3 концентрированной соляной кислоты, смесь нагревают с обратным холодильником на кипящей водяной бане в течение 15 мин, периодически встряхивая и смывая осадок со стенок. Колбу охлаждают и проводят экстракцию эфиром 3´30 см3 при нагревании на водяной бане при 35 °С с обратными холодильником в течение 15 мин с момента закипания эфира. Объединенные эфирные извлечения после охлаждения фильтруют через вату в делительную воронку на 250 см3, промывают водой 2´20 см3. При непрерывном охлаждении приливают малыми порциями сначала 15 см3 30 %-ного раствора едкого натра, затем 25 см3 щелочно-аммиачного раствора, не переставая охлаждать смесь, встряхивают 5 мин и окрашенный раствор собирают в мерную колбу на 500 см3. Экстракцию щелочно-аммиачным раствором повторяют несколько раз до прекращения окрашивания щелочного раствора. К объединенному извлечению добавляют 3 капли пергидроля, перемешивают и доводят объем щелочно-аммиачным раствором до метки (раствор 1).
Для извлечения свободных производных антрахинона 0,1 г измельченного продукта помещают в колбу на 300 см3, прибавляют 50 см3 эфира и нагревают на водяной бане с обратным холодильником 10 мин. Экстракцию повторяют трижды, эфирные извлечения объединяют, охлаждают и экстрагируют производные антрахинона 4´20 см3 щелочно-аммиачным раствором, каждый раз встряхивая по 5 мин, щелочной раствор переносят в мерную колбу на 100 см3, прибавляют каплю пергидроля, доводят объем до метки щелочно-аммиачным раствором (раствор 2).
Количественное определение антрахиноновых производных
Измеряют оптическую плотность растворов 1 и 2 при 530 нм, в качестве калибровочных используют растворы хлорида кобальта концентрациями 10 - 50 мг/см3. 10 мг/см3 хлорида кобальта соответствует концентрации свободных производных антрахинона 0,0027 мг/см3.
Содержание суммы производных антрахинона (%) вычисляют по формуле:
![]() .
.
Содержание свободных производных антрахинона (%) вычисляют по формуле:
![]() ,
где
,
где
С1 и С2 - концентрации производных анатрахинона, определенные в растворах 1 и 2,
М - масса образца,
W - потеря при высушивании, %.
Содержание связанных производных антрахинона (X, %) Х = Х1 - Х2.
20.2. В БАД, содержащих ревень тангутский (Rheum palmatum L.)
Приборы и реактивы
Спектрофотометр или ФЭК.
Щелочно-аммиачный раствор: 50 г едкого натра растворяют в 870 см3 воды, добавляют 80 см3 концентрированного раствора аммиака.
Подготовка образца
1. 1,0 г измельченного продукта помещают в плоскодонную колбу, добавляют 10 см3 10 %-ного спиртового раствора едкого натра, смесь кипятят в течение 3 - 5 мин. Раствор фильтруют, охлаждают, подкисляют разбавленной соляной кислотой до слабо кислой реакции по индикаторной бумаге, прибавляют 10 см3 эфира, взбалтывают в течение 2 - 3 мин. Эфирный слой окрашивается в желтый цвет.
2. 0,05 г продукта экстрагируют 7,5 см3 ледяной уксусной кислоты при нагревании с обратным холодильником на кипящей водяной бане в течение 15 мин. После охлаждения в колбу через обратный холодильник добавляют 30 см3 эфира и кипятят 15 мин, экстракты охлаждают, фильтруют или центрифугируют, промывают осадок на фильтре или центрифугат эфиром. Эфирные экстракты объединяют, фильтруют или центрифугируют. Осадок экстрагируют еще трижды по 10 см3 воды, объединенный экстракт фильтруют, охлаждают, фильтрат подкисляют разбавленной соляной кислотой до слабо кислой реакции по индикаторной бумаге, прибавляют 10 см3 эфира, встряхивают в течение 2 - 3 мин.
Идентификация тестовых соединений
5 см3 эфирного экстракта, полученного по п. 1, смешивают с 5 см3 раствора аммиака. Аммиачный слой приобретает вишнево-красное окрашивание (эмодины), эфирный слой остается окрашенным в желтый цвет (хризофон).
Количественное определение антраценовых производных (в пересчете на истизин)
К экстракту, полученному по п. 2, прибавляют 100 см3 щелочно-аммиачного раствора и осторожно встряхивают в течение 5 - 7 мин, охлаждая воронку под струей воды. После полного расслоения прозрачный красный нижний слой сливают в мерную колбу на 250 см3. Из эфирного слоя экстракцию продолжают аммиачно-щелочным раствором до прекращения окрашивания раствора, объединенные экстракты доводят до метки аммиачно-щелочным раствором. 25 см3 этого раствора помещают в коническую колбу на 100 см3, нагревают 15 мин. на кипящей водяной бане при периодическом перемешивании. После охлаждения раствор переносят в мерную колбу на 25 см3 и доводят объем водой до метки. Измеряют оптическую плотность раствора при 530 нм, в качестве калибровочных используют растворы хлорида кобальта 10 - 50 мг/см3. Концентрация 2,5 мг/см3 хлорида кобальта соответствует концентрации истизина 0,0009 мг/см3.
20.3. В БАД, содержащих крушину ольховидную (Frangula alnus mill)
Приборы и реактивы
Спектрофотометр или ФЭК.
Щелочно-аммиачный раствор: 50 г едкого натра растворяют в 870 см3 воды, добавляют 80 см3 концентрированного раствора аммиака.
Подготовка образца
1. 1,0 г измельченного образца помещают в плоскодонную колбу, добавляют 10 см3 10 %-ного спиртового раствора едкого натра, смесь кипятят в течение 3 - 5 мин. Раствор фильтруют, охлаждают, подкисляют разбавленной соляной кислотой до слабо кислой реакции по индикаторной бумаге, прибавляют 10 см3 эфира, встряхивают в течение 2 - 3 мин, эфирный слой окрашивается в желтый цвет.
2. 0,05 г образца экстрагируют 7,5 см3 ледяной уксусной кислоты при нагревании с обратным холодильником на кипящей водяной бане в течение 15 мин. После охлаждения в колбу через обратный холодильник добавляют 30 см3 эфира и кипятят 15 мин, экстракт охлаждают, фильтруют или центрифугируют, промывают осадок на фильтре или центрифугат эфиром. Эфирные экстракты объединяют, фильтруют или центрифугируют. Осадок экстрагируют еще трижды по 10 см3 воды, объединенный экстракт фильтруют, охлаждают, фильтрат подкисляют разбавленной соляной кислотой до слабо кислой реакции по индикаторной бумаге, прибавляют 10 см3 эфира, встряхивают в течение 2 - 3 мин.
Идентификация тестовых соединений
5 см3 эфирного экстракта, полученного по п. 1, смешивают с 5 см3 раствора аммиака. Аммиачный слой приобретает вишнево-красное окрашивание (эмодины), эфирный слой остается окрашенным в желтый цвет (хризофановая кислота).
Количественное определение антраценовых производных (в пересчете на истизин).
К экстракту, полученному по п. 2, прибавляют 100 см3 щелочно-аммиачного раствора и осторожно взбалтывают в течение 5 - 7 мин, перемешивают, охлаждая воронку под струей воды. После полного расслоения прозрачный красный нижний слой сливают в мерную колбу на 250 см3, из эфирного слоя экстрагируют аммиачно-щелочным раствором до прекращения окрашивания раствора, объединенные экстракты доводят до метки аммиачно-щелочным раствором. 25 см3 этого раствора помещают в коническую колбу на 100 см3, нагревают 15 мин. на кипящей водяной бане при периодическом перемешивании, после охлаждения раствор переносят в мерную колбу на 25 см3 и доводят объем водой до метки. Измеряют оптическую плотность раствора при 530 нм, в качестве калибровочных используют растворы хлорида кобальта 10 - 50 мг/см3. Концентрация 2,5 мг/см3 хлорида кобальта соответствует концентрации истизина 0,0009 мг/см3.
20.4. В БАД, содержащих сенну (Salvia officinalis L)
Приборы и реактивы
Спектрофотометр или ФЭК.
Щелочно-аммиачный раствор: 50 г едкого натра растворяют в 870 см3 воды, добавляют 80 см3 концентрированного раствора аммиака.
Подготовка образца
1. 0,5 г измельченного образца помещают в плоскодонную колбу, добавляют 10 см3 10 %-ного спиртового раствора едкого натра, смесь кипятят в течение 3 - 5 мин. Раствор фильтруют, охлаждают, подкисляют разбавленной соляной кислотой до слабо кислой реакции по индикаторной бумаге, прибавляют 10 см3 эфира, взбалтывают в течение 2 - 3 мин. Эфирный слой окрашивается в желтый цвет.
2. 0,4 г продукта экстрагируют 100 см3 воды при нагревании с обратным холодильником на кипящей водяной бане в течение 20 мин. После охлаждения в колбу через обратный холодильник, добавляют эфир, экстрагируют 2 раза по 30 см3, эфирные экстракты отбрасывают, водную фазу подогревают до исчезновения запаха эфира, добавляют 0,1 г натрия гидрокарбоната, 10 см3 10 %-ного раствора хлорида окисного железа, нагревают на кипящей водяной бане при перемешивании в течение 20 мин, добавляют 5 см3 50 %-ной серной кислоты и нагревают еще 20 мин. После охлаждения раствор переносят в делительную воронку, колбу ополаскивают 20 см3 воды и 75 см3 эфира, экстрагируют из объединенной водной фазы эфиром еще дважды по 20 - 30 см3, объединенные эфирные экстракты фильтруют, промывают водой. К эфирному экстракту добавляют 100 см3 щелочно-аммиачного раствора, перемешивают 5 мин., водную фазу переносят в колбу на 300 см3, к эфирному экстракту добавляют 20 см3 воды и 3 см3 концентрированной соляной кислоты, воронку охлаждают под струей воды и перемешивают слои, водную фазу переносят в колбу с водно-щелочной вытяжкой. Объединенные щелочно-аммиачные эктракты доводят до метки щелочно-аммиачным раствором.
Идентификация тестовых соединений
5 см3 эфирного экстракта, полученного по п. 1, встряхивают с 5 см3 раствора аммиака. Аммиачный слой приобретает вишнево-красное окрашивание (эмодины), эфирный слой остается окрашенным в желтый цвет (хризофановая кислота).
Количественное определение антраценовых производных (в пересчете на хризофановую кислоту).
Измеряют оптическую плотность раствора, полученного по п. 2, при 530 нм, в качестве калибровочных используют растворы хлорида кобальта 1 - 5 %; 1 %-ный раствор хлорида кобальта соответствует концентрации хризофановой кислоты 4,3 мг/дм3 щелочно-аммиачного раствора.
21. Определение гидрохинона и его производных в БАД, содержащих толокнянку (Uvae ursi folium)
Арбутин (бета-глюкозид гидрохинона) является полифенольным индикаторным компонентом толокнянки Uva Ursi. Метод анализа арбутина включает стадии кислотного гидролиза с последующим определением гидрохинона с помощью обращенно-фазовой ВЭЖХ с спектрофотметрическим детектором при длине волны 280 нм.
Приборы и реактивы
Система ВЭЖХ со спектрофотометрическим детектором.
Гидрохинон, Sigma.
Приготовление рабочего стандартного раствора
Точную навеску 1 мг стандартного образца гидрохинона переносят в мерную колбу вместимостью 100 см3, до половины содержащую метанол, встряхивают до полного растворения вещества, доводят объем метанолом до метки и тщательно перемешивают.
Подготовка образца
Содержимое капсул или растертых таблеток, содержащих около 1,0 г порошка листьев или экстракта толокнянки помещают в круглодонную колбу объемом 250 см3, добавляют 90 см3 смеси метанол - вода - концентрированная соляная кислота (70:30:2) и экстрагируют на кипящей водяной бане с обратным холодильником в течение 1 часа. Экстракт охлаждают до комнатной температуры, фильтруют через целлюлозный или тефлоновый фильтр с размером пор до 0,45 мкм в мерную колбу вместимостью 100 см3. Объем экстракта доводят дистиллированной водой до метки.
Проведение анализа.
Колонка: Kromosil C18 или аналогичная, 5 мкм, 250´4,6 мм ID;
Подвижная фаза: ацетонитрил - 0,1 М ортофосфорная кислота, рН 2,6 (20:80), скорость потока 1,0 см3/мин.
Детектирование: УФ, 280 нм.
Объем вводимой пробы 5 - 20 мкл.
Примерное время удерживания гидрохинона 4,5 - 5 мин.
Идентификацию гидрохинона проводят сравнением времени удерживания со стандартным образцом. При количественном определении используют метод внешней калибровки, сравнивая площади пиков гидрохинона в образце и стандарте.
Концентрацию арбутина определяют умножением найденной концентрации гидрохинона на коэффициент пересчета 2,473.
22. Определение производных кумарина
22.1. В БАД, содержащих крапиву (Urtica dioica L)
Скополетин (6-метокси-7-оксикумарин-гельзелиновая кислота)
Приборы и реактивы
Пластины для ТСХ «Silufol».
Облучатель ультрафиолетовый.
10 %-ный водный раствор серной кислоты.
20 %-ный раствор аммиака.
Стандартный раствор: 0,05 %-ный раствор скополетина в метаноле (раствор 1).
Подготовка образца
Экстракция стеринов
5,0 г измельченного образца помещают в плоскодонную колбу, соединенную с обратным холодильником, добавляют 100 см3 хлористого метилена, смесь нагревают на водяной бане при перемешивании в течение 30 мин. Раствор охлаждают, фильтруют, растворитель упаривают на ротационном испарителе до небольшого объема, остаток растворителя удаляют в токе воздуха. Вещество растворяют в 2 см3 хлористого метилена.
2,0 г измельченного образца помещают в плоскодонную колбу, соединенную с обратным холодильником, добавляют 100 см3 циклогексана, смесь нагревают на водяной бане при перемешивании в течение 30 мин. Смесь фильтруют, повторяют экстракцию в тех же условиях. Объединенные экстракты охлаждают, фильтруют, растворитель упаривают на ротационном испарителе до небольшого объема, остаток растворителя удаляют током воздуха. Вещество растворяют в 5 см3 смеси ацетонитрил - тетрагидрофуран (96:4).
Экстракция скополетина
5,0 г измельченного продукта помещают в плоскодонную колбу, соединенную с обратным холодильником, добавляют 50 см3 метанола, смесь нагревают на водяной бане при перемешивании в течение 30 мин. Раствор охлаждают, фильтруют, растворитель упаривают на ротационном испарителе до небольшого объема, остаток растворителя удаляют в токе воздуха. Вещество растворяют в 2 см3 метанола.
Качественный анализ скополетина с помощью метода ТСХ
На стартовую линию пластины Silufol (20´20 см), отстоящую от края пластины на 20 - 25 мм микрошприцем или стеклянным капилляром наносят в виде полосы 20´3 мм 10 мкл экстракта и 5 мкл контрольного раствора 1. Пластинку помещают в хроматографическую камеру и элюируют в смеси толуол-эфир диэтиловый (40:60), насыщенной раствором 10 %-ной уксусной кислоты. Дают фронту растворителя подняться на 10 см, затем после высушивания пластинку опрыскивают 20 % раствором аммиака. При рассматривании в УФ-свете (длина волны 360 нм) обнаруживают синюю флуоресцирующую полосу с Rf 0,25, соответствующую полосе скополетина в контрольном образце.
22.2. В БАД, содержащих вздутоплодник сибирский (Pholojodicarpus sibiricus-fisch.ex. sp re ng)
Производные пиранокумаринов: виснадин, дигидросамидин.
Приборы и реактивы
Газовый хроматограф с пламенно-ионизационным детектором.
Раствор фловерина (природная смесь дигидросамидина и виснадина) - 0,5 мг/см3 в 95 %-ном этаноле.
Подготовка образца
1 г измельченного сырья экстрагируют в аппарате сокслета хлороформом в течение 1 часа (9 - 10 сливов). Хлороформ упаривают до объема 1 - 2 см3. К остатку добавляют 5 см3 95 % этанола, содержимое количественно переносят в мерную колбу на 10 см3, объем доводят до метки 95 % этанолом.
Идентификация и количественное определение тестовых соединений
Проводят ГЖХ определение суммарного содержания производных пиранокумаринов. Условия ГЖХ: стеклянная колонка 1200´2 мм, заполненная хроматоном N-AW-HMDS с размером частиц 0,16 - 0,20 мм с 2 % лукопрена G1000.
Температура колонки - 200 °С, испарителя - 240 °С.
Скорость газа-носителя (азот) 50 см3/мин.
В инжектор вводят 5 - 6 мкл исследуемого раствора и такое же количество контрольного раствора фловерина.
Фловерин при данных условиях разделения элюируется одним пиком. Расчет производят по площадям пиков контрольного образца и пробы.
23. Определение содержания эфирных масел и подтверждение состава компонентов
Таблица 43
Компонентный состав эфирных масел (краткий перечень)
|
|
Состав эфирного масла |
|
Анис обыкновенный (Anisum vulgare Gaerth), плоды |
анетол (80 %), метилхавикол, анискетон, ацетальдегид, анисовая кислота |
|
Кориандр посевной (Coriandrum sativum L.), плоды |
d-линалоол (65 %), i-пинен, n-цимол, феландрен, дипентен, терпинолен, терпинен, гераниол, борнеол, геранилацетат, борнилацетат |
|
Эвкалипт шариковый (Eucaliptus globulus Labill), листья |
Цинеол (60 %), d-пинен, эвгенол, глобулол, пинакарнеол, сесквитерпены, альдегиды |
|
Фенхель обыкновенный (Foeniculum vulgare Mill), плоды |
Анетол (60 %), фенон (20 %) метилхавикол (10 %), пинен, камфен, диптен, фелландрен |
|
Лаванда колосовая (Lavandula spica L.), соцветия |
1-линалилацетат (35 %), спирты (10 - 30 %): 1 -линалоол, изоамиловый спирт, гераниол |
|
Мята перечная (Mentha piperita L.), листья и другие наземные части |
1-ментол (50 %), 1-ментон (10 - 20 %), эфиры ментола с уксусной, валериановой и др. кислотами (4 - 9 %) |
Приборы и реактивы
ГХ-МС с энергией ионизирующих электронов 70 эВ, квадрупольный анализатор, диапазон массовых чисел 39 - 450 а.е.м., температура источника ионов и переходных линий 200 - 250 °С. Обработка данных на МС-станции с библиотечным поиском.
ГХ с пламенно-ионизационным детектором (ГХ-ПИД).
Подготовка образца
Эфирные масла извлекают общепринятыми методами: перегонкой с водяным паром, экстракцией органическими растворителями (ГФ IX, с. 328, ГФ XI, т. 1, с. 287). Суммарное количественное определение содержания эфирных масел проводят гравиметрическим методом.
Идентификация и количественное определение состава
Анализ выделенных эфирных масел проводят с помощью методов ГХ-ПИД и ГХ-МС с использованием двух колонок разной полярности. В случае ГХ-ПИД идентификацию проводят с использованием индексов Ковача (http://www.nysaes.cornell.edu/-fst/faculty/acree/flavornet/OV101.html) при одинаковых условиях хроматографического разделения. Количественный анализ проводят с использованием метода внутреннего стандарта.
Условия хроматографического разделения (примеры)
1. ГХ-МС/ПИД: кварцевая капиллярная колонка длиной 30 - 50 м, внутренним диаметром 0,2 - 0,25 мм с неподвижной полиметилсилоксановой фазой толщиной пленки 0,2 - 0,3 мкм типа SE30 или SE54. Анализ проводят при программировании температуры: 60 °С (5 мин)®280 °С, 5 °/мин. Газ-носитель - гелий, давление на входе колонки 1 - 1,5 атм. Температура инжектора - 200 - 250 °С, температура интерфейсной линии ГХ-МС - 200 °С.
2. ГХ-МС/ПИД: кварцевая капиллярная колонка длиной 30 - 50 м, внутренним диаметром 0,2 - 0,32 мм с неподвижной полиэтиленгликолевой фазой типа ПЭГ-20М или ФФАП толщиной пленки 0,2 - 0,3 мкм. Анализ проводят при программировании температуры: 50 °С (5 мин)®220 °С, 5 °/мин. Газ-носитель - гелий, давление на входе колонки 0,5 - 1,0 атм. Температура инжектора - 200 - 220 °С, температура интерфейсной линии ГХ-МС - 200 °С.
Примечание. Из БАД на основе плодов и ягод эфирные масла предпочтительно анализировать на полярных колонках, т.к. достигается лучшее разделение основных компонентов - смешанных сложных эфиров. Эфирные масла, выделенные из БАД на основе цитрусовых и хвойных растений, содержащие терпеновые компоненты, предпочтительнее разделять на колонках SE30 и SE54.
24. Определение содержания инулина в БАД
Метод основан на определении инулина с помощью высокоэффективной жидкостной хроматографией.
Приборы и реактивы
Жидкостной хроматограф с рефрактометрическим детектором.
Смола Дауэкс 50´4, 200 - 400 меш. и Дауэкс 1´8, 200 - 400 меш.
Стандартный раствор инулина 10 мг/см3: предварительно высушивают в сушильном шкафу при 105 °С до постоянного веса в стеклянных бюксах.
Подготовка смол
Смолу Дауэкс 50´4, 200 - 400 меш. последовательно промывают на бюхнеровской воронке большими объемами 2Н NaOH, дистиллированной водой, 3Н HCl и опять водой. Избыток воды удаляют путем длительного отсасывания насосом. Готовят суспензию смолы в дистиллированной воде (1:1) и используют ее для заполнения колонки.
Ионнообменная способность для Дауэкса 50´4 в сухом виде составляет 5,1 мэкв/см3 и во влажном 1,7 мэкв/см3, соответственно.
Для работы нужна формиатная форма смолы. Последнюю готовят из смолы Дауэкс 1´8 (Сl-форма), пропуская через смолу на бюхнеровской воронке 3Н формиат натрия до тех пор, пока проба на Сl- не станет отрицательной (проба с азотнокислым серебром). После этого смолу промывают большими объемами дистиллированной воды и заполняют колонку.
Ионнообменная способность для Дауэкс 1´8 в сухом виде составляет 3,2 мэкв/см3 и во влажном виде - 1,4 мэкв/см3.
Проведение испытания
Подготовка пробы
Моно- и дисахара экстрагируют 80 % этиловым спиртом с учетом естественной влаги. Навеску образца заливают в колбе в соотношении 1:4 по сухим веществам 96 %-ным этанолом и необходимым количеством воды с расчетом, чтобы общая концентрация спирта была в пределах 80 - 82 %. Колбу нагревают с обратным холодильником на водяной бане при 70 - 80 °С в течение 15 мин. Затем спиртовую вытяжку отфильтровывают. Экстракцию повторяют в тех же условиях еще 3 - 4 раза. Спиртовые экстракты объединяют, спирт отгоняют на ротационном испарителе при температуре не выше 40 °С. Предназначенные для определения экстракты нейтральных сахаров содержат также вещества, имеющие заряд (аминокислоты, пептиды и др.). Чтобы освободить нейтральные сахара от этих примесей, экстракты пропускают через колонку со смолой Дауэкс 50´4, непосредственно соединенную с колонкой, содержащей Дауэкс 1´8. С двух колонок собирают элюат и, если необходимо, его концентрируют на роторном испарителе. Чтобы в бане не поднимать высоко температуру, воду упаривают азеотропной отгонкой с этанолом. Сконцентрированный образец количественно переносят в мерную емкость, замеряют объем и хроматографируют.
Навеску исходного образца гидролизуют 2 %-ным раствором щавелевой кислоты в течение часа при 100 °С. В качестве контроля на полноту гидролиза параллельно гидролизуют раствор инулина. Полученный после гидролиза продукт пропускают через колонки выше описанным способом, после чего хроматографически определяют суммарное количество фруктозы с помощью ВЭЖХ.
Условия ВЭЖХ
Перед работой новую хроматографическую колонку с сепароном переводят на обратнофазный режим работы. Для этого колонку промывают 40 см3 изопропанола, затем 80 см3 деионизированной воды, после чего уравновешивают колонку подвижной фазой до стабильной нулевой линии. Подвижная фаза: ацетонитрил - вода (77:23). Смесь дегазируют на роторном испарителе. Детектирование - рефрактометрическое. Скорость потока 2,5 см3/мин.
Обработка результатов
От общего количества фруктозы вычитают фруктозу моно- и дисахаров и получают количество инулина и полифруктозидов. При расчете используют коэффициент 0,9, который учитывает присоединение воды при гидролизе. Стандартный образец и испытываемую пробу хроматографируют не менее трех раз.
Ошибка метода ±2 %.
Качественный тест на инулин
Пробу обрабатывают 1 - 2 каплями 20 % спиртового раствора нафтола и 1 каплей концентрированной серной кислоты. Наблюдают красно-фиолетовое окрашивание. При замене - нафтола резорцином и тимолом, соответственно, красное и розово-малиновое окрашивание.
25. Определение содержания аралозидов А, В, С в БАД, содержащих аралию маньчжурскую (Araliae elatae (Mig) Seem)
Приборы и реактивы
Приготовление пластин для ТСХ
6 г силикагеля КСК, предварительно промытого соляной кислотой и водой и высушенного, и 0,6 г сульфата кальция растирают в ступке, добавляют 17 см3 воды и выливают на пластинку 20´20 см. Сушат в течение суток на воздухе, активируют при 120 - 140 °С 30 - 40 мин.
Стандартный раствор - 0,6 % раствор сапарала в метиловом спирте.
Подготовка образца
1. 1 г измельченного образца кипятят с 20 см3 метилового спирта на водяной бане при 80 - 85 °С с обратным холодильником в течение 1 ч, дают раствору отстояться.
2. 5 г измельченного образца помещают в аппарат сокслета с рабочим объемом 150 - 200 мл. В колбу-приемник наливают 250 мл метанола, 70 мл 50 %-ного раствора серной кислоты и проводят экстракцию на кипящей водяной бане в течение 7 ч. Полученную в приемнике смесь разбавляют водой 1:1, и колбу охлаждают под струей холодной воды в течение 10 мин. Выпавший осадок отфильтровывают, промывают на фильтре водой, подсушивают на фильтре под вакуумом. Осадок количественно переносят 50 см3 горячей смеси метилового и изобутилового спиртов (1:1,5).
Качественный тест на аралозиды
20 мкл экстракта, полученного по п. 1, и 10 мкл стандартного раствора сапарала наносят на стартовую линию пластинки с силикагелем, хроматографируют в системе хлороформ - метиловый спирт - вода (61:32:7). Дают фронту растворителя подняться на 16 - 18 см, затем после высушивания пластинку опрыскивают 20 % раствором серной кислоты и нагревают в сушильном шкафу при 105 °С 10 мин. На пластинке должны проявиться три основных пятна вишневого цвета аралозидов, соответствующих по Rf аралозидам в сапарале.
Количественное суммарное определение аралозидов
Полученный по п. 2 раствор титруют потенциометрически 0,1 М раствором едкого натра в смеси метилового спирта и бензола. При титровании отмечают количество титранта, израсходованного на доведение рН испытуемого раствора до 7,0.
Расчет суммарного содержания аралозидов в пересчете на аммонийную соль аралозидов А, В, С с усредненной молекулярной массой не менее 5 % в % на сухое вещество рассчитывают по формуле:
![]() ,
где
,
где
0,10422 - количество аммонийных солей аралозидов, соответствующих 1 см3 раствора едкого натра ОДМ;
V - объем титранта, израсходованный на титрование пробы;
V1 - объем титранта, израсходованный на доведение рН до 7,0;
V2 - объем титранта, израсходованный на холостой опыт;
W - потеря при высушивании, %;
0,2020 - коэффициент пересчета бихромата калия на гнафалозид А;
1,03 - поправочный коэффициент на неполное элюирование гнафалозида А с сорбента;
W - потеря массы при высушивании, %.
26. Определение содержания экдистена в БАД, содержащих левзею сафлоровидную (Leuzea carthamoides (Willd. DC)
Приборы и реактивы
Приготовление пластин для ТСХ: сорбент состоит из смеси Аl2О3 нейтральной и силикагеля, промытого последовательно 5Н соляной кислотой (15 - 20 ч) и водой и высушенного при 120 °С 12 час. Для приготовления пластинок 9 г силикагеля и 3 г оксида алюминия растирают в ступке с 0,4 г сульфата кальция, постепенно прибавляя 25 см3 воды, массу перемешивают и выливают на пластинку 20´20 см, высушивают и активируют при 120 °С 1 ч.
Спектрофотометр.
Раствор ГСО экдистена: 0,15 г ГСО экдистена (ВФС42-1713-87) помещают в мерную колбу на 25 см3 и доводят раствор до метки 95 %-ным этанолом. 7 см3 полученного раствора переносят в мерную колбу на 25 см3 и объем доводят до метки 95 % этанолом (раствор А).
Раствор ванилина в концентрированной серной кислоте.
Подготовка образца
15 г измельченного образца экстрагируют 180 - 200 см3 метилового спирта в аппарате Сокслета с рабочим объемом 150 см3 в течение 7 час (25 - 30 сливов). Экстракт концентрируют на ротационном испарителе при температуре 40 °С до объема 5 см3, количественно переносят в мерную колбу на 25 см3, доводят объем метанолом до метки (раствор Б). На пластинку, разделенную на 5 зон, с помощью микрошприца или стеклянного капилляра наносят в виде полос 20´3 мм 150 мкл раствора Б и 150 мкл стандартного раствора экдистена (раствор А) - на 4 зоны, пятая зона остается пустой (для раствора сравнения). Пластинку помещают в хроматографическую камеру и элюируют в смеси метанол - хлороформ - ацетон (2:6:1). Дают фронту растворителя подняться на 16 - 18 см. После высушивания пластинки две зоны опрыскивают раствором ванилина, отмечают полосу стандарта и собирают сорбент из необработанных зон стандарта и образца. Вещества смывают с силикагеля 15 мл метанола в течение 4 час.
Проведение анализа
Спектрофотометрическое количественное определение экдистена
Измеряют оптическую плотность растворов стандарта и образца относительно раствора «холостого опыта» при длине волны 242 нм в кюветах 10 мм. Содержание экдистена в сырье должно быть не менее 0,1 %.
Качественный тест на инулин
Сухой порошок обрабатывают 1 - 2 каплями 20 % спиртового раствора нафтол и 1 каплей концентрированной серной кислоты. Наблюдают красно-фиолетовое окрашивание. При замене нафтола резорцином и тимолом наблюдают, соответственно красное и розово-малиновое окрашивание.
27. Определение содержания четвертичных аммонийных оснований (глицинбетаин) в БАД, содержащих солянку холмовую (Salsola collina PALL)
Приборы и реактивы
Спектрофотометр.
ВЭЖХ с УФ-детектором.
5 % раствор соли Рейнеке (1,25 г соли Рейнеке, перекристаллизованной из воды и высушенной при 60 °С, растворяют при перемешивании в 25 см3 дистиллированной воды, нагретой до 60 °С).
Подготовка образца
1. 2 - 3 г измельченного продукта эктрагируют в Сокслете 200 см3 70 % этилового спирта в течение 2 - 2,5 ч. Экстракт фильтруют и упаривают досуха, остаток растворяют в 10 см3 1М НСl, фильтруют через бумажный фильтр, промывают 1М НСl.
Получение производного глицинбетаина для ВЭЖХ: р-бромфенациловый эфир глицинбетаина
2. Экстракт переносят в мерную колбу на 200 см3 и доводят объем 70 % этанолом. 1,25 см3 этого экстракта переносят в реакционную пробирку с пробкой на 10 см3 и растворитель упаривают досуха в токе воздуха при нагревании 80 - 90 °С. В пробирку добавляют 0,25 см3 40 % раствора ацетонитрила, содержащего 10 мМ KH2PO4, 10 мМ КНСО3, 5мМ 18-краун-6 эфира и после встряхивания добавляют 1,0 см3 раствора р-бромфенацилбромида (15 мг/см3) в ацетонитриле. После инкубации 30 мин. при 80 °С пробирку охлаждают и добавляют по 2,5 см3 0,1Н раствора серной кислоты и хлороформа. Пробирку встряхивают и центрифугируют 3 мин. Отбирают водную фазу.
Количественное определение четвертичных оснований с помощью метода спектрофотометрии
К фильтрату, полученному по п. 1, добавляют при перемешивании 5 см3 горячего 5 % раствора соли Рейнеке. Через 1 ч выпавший осадок отфильтровывают, промывают на фильтре 20 см3 эфира, переносят в стаканчик и растворяют в 15 см3 ацетона. Раствор фильтруют в мерную колбу на 25 см3, добавляют 7,5 см3 воды, доводят до метки ацетоном. Определяют оптическую плотность раствора при 520 нм. Раствор сравнения ацетон - вода (7:3).
Расчет содержания четвертичных оснований в пересчете на глицинбетаин, %, проводят по формуле:
![]() ,
где
,
где
D - оптическая плотность раствора,
153 - молекулярная масса глицинбетаинхлорида,
118 - коэффициент молярного поглощения рейнеката глицинбетаина при 520 нм,
а - навеска травы,
V - объем пробы.
Количественное определение четвертичных оснований с помощью метода ВЭЖХ
Условия хроматографии: сорбент: а) катионообменный (Nucleosil-50, SA) или б) ионно-парный (Silasorb-5, C18), размеры колонки 2´64 мм, подвижная фаза - раствор додецилсульфоната натрия, скорость потока 0,1 мл/мин. Детектор УФ-210 нм.
В хроматограф вводят 5 мл водной фазы (п. 2). Расчет - по производному глицинбетаина. Время выхода р-бромфенацилового эфира глицинбетаина - 6 мин.
28. Определение содержания гексозаминов
Для определения гексозаминов используют колориметрический медод Элсона и Моргана. (Biochem. J., 27. 1824 (1933)). Метод основан на конденсации аминосахара с щелочным ацетилацетоном с образованием 2-метил-3-ацетилпиррола и 2-метилпиррола; в присутствии реактива Эрлиха (4-диметиламинобензальдегида) 2-метилпиррол является основным хромогеном. Образование этих хромогенов сопровождается появлением красновато-розоватой окраски. В реакции этого типа глюкозамин и галактозамин дают одинаковую окраску с одинаковой величиной молярной экстинкции. Взаимодействие между сахарами и аминокислотами мешает определению, поэтому гексозамины отделяют от аминокислот на ионообменной колонке.
Если гексозамины находятся в связанном виде, то их отделяют путем гидролиза в 4Н соляной кислоте при 100 °С в течение 4 часов в запаянных ампулах под азотом.
Выделение фракции гексозаминов и отделение их от нейтральных сахаров, аминокислот и пептидов осуществляют хроматографией на смоле Дауэкс 50´4 (200 - 400 меш, Н+-форма). Смолу последовательно промывают на бюхнеровской воронке большими объемами 2Н NaOH, дистиллированной водой, 3Н HCl и опять водой. Избыток влаги удаляют путем длительного отсасывания насосом. Готовят суспензию промытой смолы в дистиллированной воде (1:1) и используют ее для заполнения хроматографических колонок.
Перед нанесением на колонку образец освобождают от соляной кислоты. Для этого его помещают в вакуумный эксикатор с NaOH. Для защиты от кислоты насос снабжают ловушкой с негашеной известью. Используют хроматографическую колонку с внутренним диаметром 10 мм (можно использовать в качестве колонки пластмассовый шприц). На дно колонки помещают маленький фильтр из стеклянной ваты, сверху наливают 5 см3 суспензии и дают ей осесть. После того, как образец проникнет в смолу, колонку промывают четырьмя порциями по 5 см3 дистиллированной воды. Гексозамины элюируют 5 - 10 см3 2Н HCl. Элюат освобождают от кислоты указанным выше способом, и в остатке сразу определяют количество гексозаминов колориметрическим методом.
Реактивы
Приготовление щелочного раствора ацетилацетона.
2,4-пентандион (ацетилацетон) должен быть бесцветным. Его накануне перегоняют и хранят в темной посуде при 4 °С. Непосредственно перед использованием 2 мл ацетилацетона растворяют в 98 см3 1М Na2CO3.
Реактив Эрлиха 2,7 %-ный (вес/объем) раствор 4-диметиламинобензальдегида в смеси этанол - концентрированная HCl (1:1, по объему). Этот реактив следует готовить в день определения.
Проведение анализа
В пробирках со стеклянными пробками смешивают 1 см3 раствора гексозамина (5 - 30 мкг) с 1 см3 раствора щелочного ацетилацетона. Пробы нагревают в течение 45 мин при 100 °С и затем охлаждают водопроводной водой. Добавляют 4 см3 95 %-ного этанола и тщательно перемешивают, а затем добавляют 1 см3 реактива Эрлиха и опять интенсивно перемешивают. На последнем этапе следует периодически выпускать образующийся СО2, приоткрывая пробку несколько раз во время перемешивания. Пробирки оставляют на 1 ч при комнатной температуре и затем определяют оптическую плотность раствора при 530 нм. Параллельно проводят обработку стандартных и слепых проб.
29. Определение содержания гуминовых кислот и глицина в мумие
Методика основана на гравиметрическом определении содержания гуминовых кислот, экстрагированных из продукта щелочным раствором. Глицин идентифицируют в кислотном гидролизате методом ТСХ при обнаружении пятен аминокислот обработкой нингидроновым реагентом.
Приборы и реактивы
Спектрофотометр.
1 % раствор NaOH.
6Н, 0,5Н растворы НСl.
0,3 % раствор нингидрина в н-бутаноле.
Уксусная кислота концентрированная.
25 % раствор аммиака.
Раствор глицина в 0,5H НСl - 2 мг/см3.
Подготовка образца
К 2 г образца добавляют 25 см3 1 % раствора NaOH и нагревают в колбе с обратным холодильником в течение 2 - 2,5 ч на кипящей водяной бане. Экстракт фильтруют или центрифугируют при 3000 об./мин., остаток снова обрабатывают 10 см3 раствора щелочи. Объединенные экстракты фильтруют или центрифугируют, фильтрат подкисляют 5 % НСl до рН 3 - 4, колбу с выпавшим осадком помещают на 30 - 60 мин в холодильник, далее взвесь переносят в ц/ф пробирку с известным весом, центрифугируют, осадок промывают водой, снова центрифугируют, супернатант отделяют, а осадок высушивают при температуре 105 °С до постоянного веса. В образце определяют содержание золы, прокалив его при температуре 350 °С в течение 5 - 6 ч. Содержание беззольных гуминовых кислот рассчитывают по разнице массы гуминовых кислот и золы. Записывают спектры раствора гуминовых кислот с концентрацией 2 мг/см3 в 0,1Н NaOH в области длин волн 465 - 730 нм. Раствор гуминовых кислот мумие характеризуется максимами с убывающей интенсивностью при 726, 665, 620, 575, 530, 500, 465 нм.
200 мг образца помещают в запаяную стеклянную ампулу, добавляют 10 см3 6Н НСl и проводят гидролиз при 100 - 105 °С в течение 24 ч; далее кислоту упаривают досуха. Остаток растворяют в 0,5Н НСl.
Идентификация глицина
На стартовую линию пластины Силуфол наносят по 2 - 3 мкл растворов образца и глицина и помещают в хроматографическую камеру, содержащую смесь бутанола, уксусной кислоты и воды, взятых в объемном соотношении 4:1:1. Дают растворителю поднятся на 10 см, пластинку вынимают, сушат от растворителя и опрыскивают 0,3 % раствором нингидрина. Помещают пластину на 5 - 10 мин в термостат при 105 °С. Глицин обнаруживают в виде красного пятна с Rf 0,5.
Глава 4.
МЕТОДЫ ОПРЕДЕЛЕНИЯ ПИЩЕВЫХ ДОБАВОК В СОСТАВЕ БАД
I. Метод определения консервантов (бензойная и сорбиновая кислоты) с помощью ВЭЖХ
Сущность метода
Метод основан на извлечении бензойной кислоты (БК) и сорбиновой кислоты (СК) из БАД перегонкой с паром и/или экстракцией органическим растворителем с последующим хроматографическим разделением их в тонком слое сорбента, элюции и измерении оптической плотности полученных элюатов.
Подготовка к испытанию
Приготовление стандартных растворов
Раствор 1. Навеску 100 мг бензойной кислоты переносят в мерную колбу на 25 см3 и доводят до метки этилацетатом (концентрация полученного раствора 4 мг/см3).
Раствор 2. Навеску 40 мг сорбиновой кислоты переносят в мерную колбу на 100 см3 и доводят до метки этилацетатом (концентрация полученного раствора 0,4 мг/см3).
Раствор 3. Смешивают равные объемы растворов 1 и 2. Концентрация БК в полученном растворе 2,0 мг/см3, СК - 0,2 мг/см3.
Выделение бензойной и сорбиновой кислот
Анализируемую пробу продукта (кроме напитков) массой около 10 г отвешивают с точностью до 0,01 г, измельчают и гомогенизируют с добавкой 25 г Na2SO4 и 40 см3 1М H2SO4. Гомогенат переносят в колбу емкостью 1 дм3, соединенную с парообразователем и нагревают (рис. 25).
В момент, когда жидкость в колбе начинает закипать, закрывают парообразователь пробкой и отгоняют БК и СК с паром, собирая около 80 см3 дистиллята в приемник, содержащий 10 см3 1М NaOH. Дистиллят переносят в делительную воронку, насыщают Na2SO4 (на 10 см3 дистиллята добавляют 6 г Na2SO4), подкисляют 1М H2SO4 до рН 2,0 - 3,0 и экстрагируют этилацетатом трижды по 10 см3. Объединенный экстракт сушат, добавляя 2 г прокаленного безводного Na2SO4. Этот экстракт обозначают V1. Экстракт упаривают на ротационном испарителе (допускается упаривание в фарфоровой чашке на песчаной бане) до объема 1 см3.
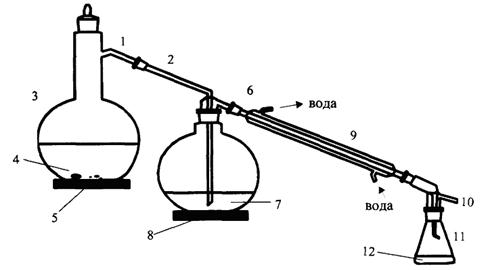
Рис. 25. Прибор для перегонки с паром.
1- пробка парообразователя; 2 - насадка парообразователя; 3 - парообразователь с дистиллированной водой; 4 - кипелки; 5 - нагреватель парообразователя; 6 - насадка; 7 - гомогенат продукта; 8 - нагреватель; 9 - холодильник; 10 - алонж; 11 - приемная колба; 12 - раствор щелочи.
При анализе напитков исключают стадию отгонки, 10 см3 напитка разбавляют вдвое 0,5М H2SO4, добавляя 10 г Na2SО4, интенсивно перемешивают и экстрагируют БК и СК 3 раза по 5 см3 этилацетатом. Объединенный экстракт (V1) сушат 1 г безводного прокаленного Na2SО4. Экстракт упаривают на ротационном испарителе или в фарфоровой чашке до конечного объема 1 см3.
Количественное определение с помощью ВЭЖХ согласно ГОСТ Р 30059-93 «Напитки безалкогольные. Методы определения аспартама, сахарина, кофеина и бензоата натрия».
Хроматографический анализ
Аппаратура, материалы
Жидкостной хроматограф с УФ детектором, длина волны 272 нм.
Автоматическая система обработки данных типа «Амперсент» или интегратор.
Колонка «Kromasil 5m С18», размер колонки 250´4,6 мм, размер частиц 5 мкм.
Полученный экстракт и раствор стандарта 3 поочередно вводят в жидкостный хроматограф при следующих условиях работы: состав элюэнта 0,025 М ацетат натрия, рН 4,5-ацетонитрил (8:2), скорость подвижной фазы 1,0 см3/мин, детектирование в УФ-спектре при 272 нм, объем вводимой пробы 5 - 10 мкл, чувствительность детектора 0,01 О.Е.П.
На хроматограмме экстракта идентифицируют пики БК и СК по времени удерживания стандартов: для БК - 3,4 мин ±0,2, для СК - 4,4 ±0,2 мин.
Для подтверждения правильности идентификации консервантов аликвоты раствора 3 и экстракта вводят в хроматограф в тех же условиях повторно, регистрируя в максимумах хроматографических пиков спектр поглощения БК (250 - 270 нм) и СК (230 - 270 нм). БК имеет три максимума поглощения в указанном интервале спектра -268, 270 и 272 нм, а СК - один - 254 нм, что является дополнительным идентификационным признаком для подтверждения присутствия их в образце.
Обработка результатов
Измеряют высоту пиков стандартов БК и СК на хроматограммах и рассчитывают их содержание в мг/кг или мг/дм3 по формуле:
![]() ,
где
,
где
К - коэффициент, учитывающий степень извлечения БК и СК;
Р - концентрация БК или СК в растворе стандарта (раствор 3), мг/см3;
Н0 - высота пика БК или СК на хроматограмме экстракта, мм;
Нст - высота пика БК или СК на хроматограмме стандартов, мм;
V1 - объем экстракта, см3;
М - масса образца, взятая для анализа (г) или объем напитка (см3).
Вычисление проводят до второго десятичного знака.
За конечный результат испытаний принимают среднее арифметическое двух параллельных определений.
Окончательный результат округляют до первого десятичного знака.
Метрологические характеристики
Относительное допустимое расхождение между результатами двух параллельных определений, выполненных в одной лаборатории, по отношению к среднему арифметическому значению (Rr) и относительное допустимое расхождение между результатами испытаний, выполненных в двух разных лабораториях, по отношению к среднему арифметическому значению (RR) приведены в табл. 44.
Таблица 44
Относительные допустимые внутрилабораторные (Rr) и межлабораторные (RR) расхождения результатов определения
|
Содержание, мг/кг |
Бензойная кислота |
Сорбиновая кислота |
||
|
Rr, % |
RR, % |
Rr, % |
RR, % |
|
|
150 - 300 |
15 |
30 |
15 |
30 |
|
300 - 2000 |
15 |
25 |
15 |
25 |
II. Метод определения заменителей сахара
Идентификация и количественное определение осуществляется методом высокоэффективной жидкостной хроматографии.
1. Метод определения аспартама
Аспартам: 1-Метиловый эфир N-L-a-аспартил-L-фенилаланина.
Условия ВЭЖХ
Колонка из нержавеющей стали, длина 150 мм, диаметр 4,6 мм. Сорбент-Кромасил 100 С18, зернение 5 мкм. Перед работой новую колонку необходимо перевести на обратнофазный режим работы. Для этого колонку промывают 40 см3 изопропанола, затем 80 см3 деионизированной воды, после чего уравновешивают колонку подвижной фазой до стабильной нулевой линии.
Подвижная фаза
5,6 г фосфорнокислого калия однозамещенного, безводного (КH2РO4) растворяют в 820 см3 бидистиллированной воды, доводят рН до 4,3 добавлением фосфорной кислоты, после чего прибавляют 180 см3 ацетонитрила. Смесь дегазируют на роторном испарителе.
Стандарт аспартама с концентрацией 1 мг/см3.
Детектирование
Спектрофотометр, длина волны УФ 255 нм, шкала 0,5 AUFS, скорость потока 0,8 см3/мин., время удерживания аспартама - 6,2±0,05 мин. Обработка хроматографических данных по программе МультиХром для Windows версия 1.39.
Стандартный образец и испытываемую пробу хроматографируют не менее трех раз. Ошибка метода ±5 %.
2. Метод определения дикетопиперазина
Дикетопиперазин: 5-Бензил-3,6-диокси-2-пиперазилуксусная кислота.
Подготовка образца
При определении аспартама и дикетопиперазина в БАД учитывают неустойчивость аспартама к нагреванию, а также то, что он гидролизуется в сильнокислых и нейтрально-щелочных средах.
Проведение анализа
Подготовка хроматографической колонки и условия ВЭЖХ - по методу 1.
Детектирование: УФ 210 нм, шкала 0,01 AUFS, скорость потока 0,8 см3/мин, стандартный раствор 2 мкг/см3, время удерживания дикетопиперазина 3,9±0,02. Ошибка метода ±2 %.
3. Метод определения ацесульфама К
Ацесульфам: 6-метил-1,2,3-оксатиазин-4 (3Н)-он-2,2-диоксид, калиевая соль.
Проведение анализа
Подготовка хроматографической колонки и условия ВЭЖХ - по методу 1.
Детектирование: УФ 225 нм, шкала 0,05 AUFS, время удерживания 2,8±0,02.
4. Метод определения сахарина
Сахарин: 3-оксо-2,3-дигидробензо[d]изотиазол-1,1-диоксид.
Подготовка образца
При выделении из БАД учитывают следующие свойства сахарина: 1 г сахарина растворяется в 290 см3 холодной воды или в 25 см3 кипящей воды, в 31 см3 этилового спирта и в 12 см3 ацетона. Сахарин практически нерастворим в хлороформе, хорошо экстрагируется этиловым и петролейным эфирами. Растворимость его увеличивается при добавлении лимонной, винной или уксусной кислот. Он устойчив при нагревании до 150 °С при рН 3,3 и 7,0.
Проведение анализа
Подготовка хроматографической колонки и условия ВЭЖХ - по методу 1.
Детектирование: УФ 269 нм, шкала 0,1 AUFS, время удерживания 2,9±0,1 мин.
5. Метод определения цикламата и цикламата в смеси с сахарином
Цикламат: натриевая соль циклогексиламино-N-сульфоновой кислоты.
Условия ВЭЖХ
Колонка из нержавеющей стали, длина 150 мм, диаметр 4,6 мм. Сорбент-Кромасил 100 С18, зернение 5 мкм. Перед работой новую колонку необходимо перевести на обратнофазный режим работы согласно описанию в методе 1.
Подвижная фаза
5,6 г фосфорнокислого калия однозамещенного, безводного (KH2PO4) растворяют в 850 см3 бидистиллированной воды, доводят рН до 1,8 добавлением фосфорной кислоты, после чего прибавляют 150 см3 ацетонитрила. Смесь дегазируют на роторном испарителе.
Детектирование
Спектрофотометр, длина волны УФ 210 нм, шкала 0,02 AUFS, скорость потока 0,6 см3/мин, время удерживания цикламата - 7,1±0,02 мин, сахарина - 8,5±0,05 мин.
Стандартный образец и испытываемую пробу хроматографируют не менее трех раз. Ошибка метода ±5 %.
6. Метод определения сукралозы
Сукралоза: 1,6-дихлор-1,6-дидеокси-b-D-фруктофуранозил-4-деокси-a-D-галактопиранозид; 4, 11, 61-трихлоргалактосахароза.
Условия ВЭЖХ
Колонка из нержавеющей стали, длина 150 мм, диаметр 4,6 мм. Сорбент: сепарон SGX С18, зернение 7 мкм. Перед работой новую колонку необходимо перевести на обратнофазный режим работы согласно описанию в методе 1.
Подвижная фаза
Ацетонитрил - вода (15:85).
Стандарт концентрацией 1,0 мг/см3. Стандартный раствор готовят в подвижной фазе.
Детектирование: рефрактометр, скорость потока 0,7 см3/мин, время удерживания сукралозы - 6,5±0,05 мин. Ошибка метода ±5 %.
7. Метод определения изомальта
Пищевая добавка, заменитель сахара Е 953 изомальт - дисахаридный полиол, состоящий из смеси примерно равных частей D-глюкопиранозил-1,6-D-сорбита (GPS) и D-глюкопиранозил-1,1-D-маннита (GPM). Брутто формула С12H24O11.
Принципиальная схема метода
Метод заключается в количественном определении основных компонентов изомальта (GPS и GPM) или суммы их концентраций и предусмотренных спецификацией примесей сорбита, маннита, изомальтулозы, а также глюкозы, фруктозы и сахарозы в исследуемом экстракте с помощью катионообменной ВЭЖХ в форме Са++ при температуре хроматографической колонки 80 °С или с помощью ВЭЖХ на аминофазе с использованием рефрактометрического детектора.
Материалы и реактивы
Жидкостной хроматограф с насосом высокого давления с подачей растворителя от 0,1 до 5 см3/мин. с устройством для термостатирования колонки.
Колонка и предколонка с катионитом в форме Са++ «НРХ-87 С, BioRad» или аналогичная колонка и предколонка на основе сульфированного стиролдивинилбензола в ионной кальциевой форме (8 %), например, «Rezex RCU-USP Sugar Alcohols» или «Waters Sugar-Pak», длина колонки 250 мм, предколонки - 30 мм, внутренний диаметр колонки и предколонки 4,0 или 4,6 мм;
Колонка и предколонка Kromasil NH2 (MetaChem Technologies, США) или аналогичная, параметры колонки 250´4,6 мм, размер частиц сорбента 5 мкм;
Рефрактометрический детектор; например, MERK L 7490, Германия, регистрирующий потенциометр (самописец) или интегратор или автоматическая система обработки данных «МультиХром для Windows 9x&NT», версия 1,5х;
Весы лабораторные общего назначения по ГОСТ 24104 с допустимой погрешностью ±0,01 г;
Весы лабораторные общего назначения с допустимой погрешностью ±0,001 г.;
Микрошприцы МШ-10 и МШ-25 для жидкостной хроматографии;
Вакуум-сушилка;
Эксикатор;
Термометр ртутный стеклянный лабораторный на 200 °С по ГОСТ 215-73;
Изомальт фирмы Sudzucker, LIMS No 200009040;
Фруктоза, глюкоза, сорбит, маннит, сахароза, изомальтоза кристаллические;
Аппарат для встряхивания типа АВУ-6С по ТУ 64-1-2451-78;
Капроновый фильтр марки 0,45 мкм RC;
Вода бидистиллированная деионизированная по ГОСТ 6709 и фильтрованная через капроновый фильтр марки 0,45 мкм RC;
Шкаф сушильный, обеспечивающий нагрев до 150 °С;
Бумажные фильтры обеззоленные марки ФОМ;
Бюкса с притертой крышкой по ГОСТ 7148-70;
Колбы мерные по ГОСТ 1770 вместимостью 100 и 250 см3;
Цинк уксуснокислый 2-водный, х.ч. по ГОСТ5823-78; раствор концентрацией 300 г/дм3.
Приготовление стандартных растворов
Аналитический образец изомальта высушивают до постоянного веса согласно ГОСТ 12570-67 «Сахар-песок и сахар-рафинад. Метод определения содержания влаги». Готовят стандартный раствор изомальта с массовой концентрацией 3 мг/см3. Точные концентрации GPS и GPM в полученном стандартном растворе рассчитывают, исходя из навески изомальта и количеств каждого компонента смеси, декларированных в аналитическом сертификате. Таким образом, концентрации GPS и GPM должны составить от 1,4 до 1,6 мг/см3.
Готовят стандартные растворы фруктозы, глюкозы, сорбита, маннита, сахарозы, изомальтулозы с массовыми долями 3 мг/см3.
Приготовление исследуемого образца
Анализируемый образец БАД или пищевого продукта размельчают или растирают в ступке. Навеску массой 5±0,01 г помещают в коническую колбу вместимостью 250 см3, добавляют 150 см3 подогретой до 75 - 80 °С дистиллированной воды и встряхивают на аппарате для встряхивания 20 - 30 минут. Доводят температуру смеси до комнатной, переносят в делительную воронку, встряхивают с хлороформом (3 раза по 50 см3), хлороформный слой отбрасывают, верхний водный слой количественно переносят в мерную колбу вместимостью 250 см3. Добавляют 15 см3 30 %-ного раствора ацетата цинка, встряхивают, доводят дистиллированной водой до метки и тщательно перемешивают. Фильтруют через капроновый фильтр марки 0,45 мкм RC или складчатый бумажный фильтр «синяя лента». Остаток хлороформа из фильтрата отдувают в токе азота и аликвоту вводят в хроматограф.
Анализ с помощью ВЭЖХ
I вариант (с использованием катионообменной ВЭЖХ).
Условия ВЭЖХ: температура 80 - 90 °С, подвижная фаза бидистиллированная, деионизированная и фильтрованная вода, скорость подвижной фазы 0,4 - 0,8 см3/мин., аликвота, вводимая в инжектор 10 - 15 мкл; чувствительность рефрактометрического детектора устанавливается таким образом, чтобы ввод 1,5 мкг GPS или GPM соответствовал отклонению пера на полную шкалу самописца при уровне шума от 1 до 3 % от полной шкалы; входное напряжение регистрирующего потенциометра (самописца) или интегратора 10 mV.
Определяют каждый компонент изомальта (GPS и GPM) и суммируют их.
Ориентировочный порядок времен удерживания углеводов в минутах при использовании катионообменной ВЭЖХ (зависят от типа колонки, скорости подвижной фазы и температуры колонки): сахароза - 9±1, изомальтулоза - 10±1, глюкоза - 12±2, GPM - 14±1, фруктоза - 15±1, GPS - 16 ± 1, маннит - 22±2, сорбит - 29±2.
II вариант (с использование аминофазы).
Условия ВЭЖХ: подвижная фаза ацетонитрил - вода (77:23), скорость подвижной фазы 1,0 см3/мин., аликвота, вводимая в инжектор 10 - 15 мкл; чувствительность рефрактометрического детектора устанавливают таким образом, чтобы ввод 30 мкг фруктозы, глюкозы или сахарозы соответствовал отклонению пера на полную шкалу самописца при уровне шума от 1 до 3 % от полной шкалы; входное напряжение регистрирующего потенциометра (самописца) или интегратора 10 mV.
Определяют сумму концентраций (GPS и GPM) в виде изомальта.
Ориентировочный порядок времен удерживания углеводов в минутах при использовании амино-фазы: фруктоза - 7,0±0,2, сорбит 7,5±0,2, глюкоза 8,5±0,1, сахароза 11±0,1, изомальт 13±0,2.
Метрологические характеристики метода
Предел обнаружения метода по манниту, сорбиту и изомальтулозы - около 30,0 мг/дм3 или 0,003 %. Стандартное отклонение сходимости (Sr) при концентрации сорбита 1 % не превышает 0,1 - 0,13. Среднее значение открываемости маннита, сорбита и изомальтулозы 91 - 95 %, GPS и GPM - 96 - 98 %.
Метод позволяет определять содержание GPS, GPM или изомальта, а также концентрации примеси глюкозы, фруктозы, сорбита, маннита, сахарозы и изомальтозы.
Обработка результатов измерений
Количественное определение компонентов осуществляют методом внешнего стандарта. Для калибровки прибора в инжектор с помощью микрошприца вводят 0,005, 0,01 и 0,02 см3 соответствующих стандартных растворов. Для каждого количества определяют площадь пика на хроматограмме.
В инжектор хроматографа вводят 0,01 см3 (10 мкл) раствора анализируемого образца. Определяют площадь пика, соответствующего по времени удерживания стандартам. Расчет концентрации компонентов проводят по формуле:
![]() ,
%, где
,
%, где
С - концентрация компонента в образце, %;
V1 - объем анализируемого образца, см3 (250 см3);
V2 - объем аликвоты раствора анализируемого образца, внесенный в хроматограф, (0,01 см3);
m - масса стандарта, введенного в хроматограф при калибровке, мкг;
М - навеска образца, взятая для анализа, г;
![]() - площадь пика в анализируемом образце;
- площадь пика в анализируемом образце;
![]() - площадь пика стандарта.
- площадь пика стандарта.
III. Методы определения состава ароматизаторов
Состав и содержание ароматизаторов определяют методом хромато-масс-спектрометрии (ХМС). Если допускает состав пробы, ХМС-анализ проводят по следующим схемам:
а) прямой ХМС анализ исходного образца;
б) ХМС анализ экстракта.
Приборы
Хромато-масс-спектрометр с автоматической системой обработки данных и библиотекой масс-спектров.
Подготовка пробы
Органические компоненты экстрагируют из образца, разбавленного водой в соотношении 1:5, смесью эфир-пентан (5´3 см3); если растворитель - пропиленгликоль, экстракцию проводят пентаном. Экстракт обезвоживают сульфатом натрия и упаривают в токе азота при комнатной температуре до 100 мкл, 1 мкл экстракта вводят в инжектор.
Условия хроматографического разделения
Кварцевая капиллярная колонка длиной 30 - 50 м, внутренним диаметром 0,2 - 0,23 мм, неподвижная фаза полиметилсилоксан типа SE 30, толщина пленки 0,2 - 0,3 мкм, газ-носитель гелий, давление на входе колонки 1,6 атм. Температура источника ионов 160 °С, испарителя и переходных линий 220 и 200 °С, режим программирования температуры колонки: 50 °С (2 мин), 120 °С (5 мин), далее - повышение температуры за 4 мин до 250 °С, изотерма - 20 мин.
Условия масс-спектрометрии: энергия ионизирующих электронов 70 eV, сканирование по ПИТ от 39 до 450 а.е.м., обработка данных с использованием библиотек масс-спектров для идентификации отдельных ароматических компонентов (http://www.nysaes.cornell.edu/fst/faculty/acree/flavornet/OV101.html).
Список разрешенных ароматизаторов приведен в СанПиН 2.3.2.1293-03 «Продовольственное сырье и пищевые продукты. Гигиенические требования по применению пищевых добавок».
IV. Метод определения синтетических пищевых красителей
К синтетическим пищевым красителям относятся органические соединения следующих групп: азокрасители, пиразолоновые, трифенилметановые, антрахиноновые, индигоидные, ксантеновые, хинолиновые и полициклические (Е102 - «Тартразин», Е103 - «Алканет», Е104 - «Желтый хинолиновый», Е107 - Желтый 2G, Е110 - «Желтый солнечный закат», Е122 - «Азорубин, Кармуазин», Е124 - «Понсо 4R», Е128 -«Красный 2G», Е129 - «Красный очаровательный AC», E131 - «Синий патентованный FCF», E142 - «Зеленый S», E143 - «Зеленый прочный FCF», E151 - «Черный блестящий PN», E155 - «Коричневый NT»).
В зависимости от типов заместителей растворимость в воде рассматриваемых красителей колеблется от очень хорошей до очень плохой. Увеличение числа сульфатных или карбоксильных групп повышает растворимость в воде. Наличие заместителей (хлор, нитрогруппа или метил) повышает растворимость красителей в органических растворителях. В настоящей методике органические красители разделены на несколько групп в соответствии с их химическим строением: азокрасители, пиразолоновые, трифенилметановые, антрахиноновые, индигоидные, ксантеновые, хинолиновые и полициклические.
Таблица 45
Перечень и свойства синтетических пищевых красителей
|
Краситель |
Номер по CI |
Индекс FD&C |
Макс. поглощения, нм |
Коэффициент светопоглощения эталона (Э) |
|
1 |
2 |
3 |
4 |
5 |
|
Тартразин Е102 |
19140 |
FD&C Yellow 5 |
427 |
0,053 |
|
Алканет Е103 |
14270 |
- |
425 |
0,064 |
|
Хинолиновый желтый Е104 |
47005 |
Food Yellow 13 |
412 |
0,096 |
|
Желтый «Солнечный закат» Е110 |
15985 |
Food yellow 6 |
484 |
0,054 |
|
Кармины Е120 |
75470 |
Natural Red 4 |
|
|
|
Азорубин Е122 |
14720 |
Food Red 3 |
228 |
0,045 |
|
Амарант Е123 |
16189 |
Food Red 9 |
521 |
|
|
Понсо 4R Е124 |
14700 |
- |
502 |
0,054 |
|
Эритрозин Е127 |
45430 |
FD&C Red 3 |
525 |
0,057 |
|
Красный 2G E128 |
18050 |
Food Red 10 |
|
|
|
Красный «Очаровательный» Е129 |
16035 |
- |
500 |
0,052 |
|
Патентованный синий Е131 |
42051 |
- |
639 |
0,048 |
|
Индигокармин Е132 |
73015 |
FD&C Blue 2 |
610 |
0,048 |
|
Синий блестящий FCF Е133 |
42090 |
FD&C Blue 2 |
630 |
0,164 |
1. Определение качественного состава красителей в БАД методом ТСХ
1.1. Качественное определение индивидуальных и смесевых синтетических пищевых красителей
Качественный состав индивидуальных и смесевых пищевых красителей (СПК) определяют методом ТСХ на силикагеле в системах:
Система А
н-пропанол 100 мл
этилацетат 30 мл
вода 30 мл
аммиак 1 мл
Система В
уксусная кислота 20 мл
изобутанол 50 мл
вода 20 мл
Наносят на пластинку ТСХ 2-5 мкл раствора исследуемого образца с концентрацией 10 мг/100 см3. Проводят разделение в системе А.
Таблица 46
Величины Rf некоторых СПК при ТСХ
|
Наименование красителя |
Величина Rf (система А) |
Величина Rf (система В) |
|
1 |
2 |
3 |
|
Тартразин Е102 |
0,595 |
0,19 |
|
Желтый хинолиновый Е104 |
|
0,56 |
|
Желтый «Солнечный закат» Е110 |
0,690 |
0,57 |
|
Кармуазин Е122 |
0,78 |
0,51 |
|
Понсо 4R E124 |
0,571 |
0,51 |
|
Эритрозин Е127 |
|
0,97 |
|
Красный «Очаровательный» Е129 |
|
- |
|
Синий патентованный Е131 |
|
0,5 |
|
Индигокармин Е132 |
0,714 |
0,26 |
|
Синий блестящий FCF E133 |
|
0,5 |
|
Зеленый S E142 |
0,571 |
0,35 |
|
Зеленый прочный FCF E143 |
|
0,18 |
В случае, если в системе А смеси пищевых красителей не разделились, либо величины Rf пищевых красителей, находящихся в смеси, близки проводят хроматографическое разделение в системе В, либо двумерную ТСХ, используя в качестве второй подвижной фазы систему В.
1.1.1. Очистка образца
В случае, если в матриксе пищевого красителя присутствуют вещества, препятствующие нанесению на пластинку ТСХ, либо способствующие необратимой сорбции образца пищевого красителя на старте, необходимо провести предварительную очистку образца. Для этого на стеклянный фильтр диаметром 25 - 30 мм помещают целлюлозу в виде суспензии в 25 %-ном растворе сульфата аммония слоем толщиной около 5 мм. На слой целлюлозы наносят очищаемый образец и производят элюирование поэтапно: метанолом (20 см3) и 25 %-ным раствором сульфата аммония (80 см3) до исчезновения окраски целлюлозы. Полученный элюат используют для проведения анализа методом ТСХ.
1.1.2. Очистка образца хроматографическими методами
10 - 20 г порошка целлюлозы перемешивают примерно с 200 см3 воды. Дают смеси отстояться и декантируют жидкость. Повторяют промывку и декантацию. К промытой целлюлозе добавляют приблизительно 100 см3 элюента, перемешивают и переносят суспензию в хроматографическую колонку. После стекания жидкости промывают содержимое колонки еще 100 см3 элюента. 5 г целлюлозы промывают, как описано выше. К промытой целлюлозе добавляют 100 см3 элюента, перемешивают, дают смеси отстояться и отделяют жидкость декантацией.
Соединяют 5 г промытой целлюлозы с образцом красителя, добавляют 10 г сульфата аммония. Тщательно перемешивают. Количественно переносят смесь в колонку. Для ополаскивания используют приблизительно 25 см3 элюента. Когда весь раствор стечет, добавляют около 400 см3 элюента. Собирают восемь фракций по 50 см3 каждая и анализируют с помощью ТСХ.
1.2. Количественное определение состава синтетических пищевых красителей в БАД с помощью ТСХ и спектрофотометрии
1.2.1. Обработка образца
Водорастворимые БАД
Твердый образец
Навеску 5,0 г образца и переносят в плоскодонную колбу вместимостью 250 см3. Добавляют примерно 50 см3 кипящей воды и встряхивают до полного растворения образца. Переносят окрашенные соединения на полиамидный порошок согласно п. 1.2.2.
Жидкий образец
Отбирают 50,0 см3 образца и переносят окрашенные соединения на полиамидный порошок согласно п. 1.2.2.
БАД с высоким содержанием жира
Обезжиривают образец при помощи петролейного эфира. Навеску 5,0 г переносят в центрифужную пробирку. Добавляют приблизительно 20 см3 петролейного эфира и перемешивают при помощи стеклянной палочки. Декантируют петролейный эфир. Повторяют до полного обезжиривания. Если образец не содержит жира, его переносят в плоскодонную колбу вместимостью 250 см3, добавляют 50 см3 кипящей воды и далее действуют согласно п. 1.2.2.
БАД с высоким содержанием крахмала и белка
Навеску 5,0 г БАД переносят в центрифужную пробирку. Добавляют 25 см3 кипящей воды и перемешивают. Добавляют 25 см3 смеси метанол - 5 %-ный аммиак 95:5. Проверяют рН (величина рН должна составлять приблизительно 9). Тщательно перемешивают. Оставляют образец в холодильнике примерно на 15 мин. Центрифугируют при 2000 об/мин в течение 10 мин. Декантируют чистый раствор в плоскодонную колбу емкостью 250 см3. Добавляют 5 см3 воды в центрифужную пробирку, перемешивают и добавляют 10 см3 смеси метанол - 25 %-ный аммиак 95:5. Перемешивают и центрифугируют, как описано выше. Повторяют указанную процедуру до полной экстракции окрашенных соединений. Объединяют все экстракты. Упаривают объединенный экстракт на водяной бане приблизительно до объема 25 см3 (чтобы удалить весь метанол). Добавляют 25 см3 воды и нагревают раствор. Далее действуют согласно п. 1.2.2.
1.2.2. Перевод красителей на полиамидный порошок
Используя раствор уксусной кислоты в метаноле (1:1 по объему) или водный аммиак доводят рН образца до 4 - 5. Добавляют 1 г полиамидного порошка к теплому раствору. Перемешивают в течение 1 минуты. Дают порошку осесть. Проверяют исчезновение окраски раствора. Если окраска сохранилась, добавляют еще некоторое количество полиамидного порошка и перемешивают. (Синтетические красители полностью абсорбируются на полиамиде, натуральные красители - лишь частично. Свойство красителей абсорбироваться на полиамидной матрице можно использовать для определения наличия натурального красителя в образце). Полиамидный порошок: SC6 «Machery Nagel & Со» Duren, размер пор 0,05 - 0,16 мм.
Перемешивают и переносят суспензию на стеклянную колонку. Промывают плоскодонную колбу тремя порциями по 10 см3 горячей воды и переносят на колонку. Колонку промывают 10 см3 горячей воды и затем 3´5 см3 метанола. Стеклянная колонка: длина 20 см, внутренний диаметр - 30 мм.
1.2.3. Элюирование и концентрирование индивидуальных красителей
Помещают круглодонную колбу вместимостью 100 см3 под колонку и элюируют красители с полиамида растворителем (метанол - 25 %-ный аммиак 95:5 по объему) порциями по 5 мл со скоростью 2 мл/мин до тех пор, пока полиамид не обесцветится. Упаривают элюат досуха на вакуумном испарителе при температуре бани не выше 40 °С. Добавляют 1 - 2 см3 смеси метанол - 25 %-ный аммиак 95:5. Используют полученный раствор для разделения красителей методом ТСХ.
1.2.4. Разделение методом ТСХ
Используя микрошприц или микропипетку, наносят на хроматографическую пластинку (целлюлоза, толщина слоя 0,10 мм, DC-Fertigplatten, Merck Art 5716 без флуоресцентного индикатора) полосу образца, полученного по п. 1.2.3 длиной около 3 см. Объем пробы 50 - 100 мкл. Высушивают нанесенный образец феном.
ТСХ проводят в одной из трех систем растворителей:
2,5 %-ный водный раствор цитрата натрия (C6H5Na3O7´2H2O) - 25 %-ный аммиак - метанол, 80:20:12 (по объему)
1-пропанол - этилацетат - вода, 6:1:3 (по объему)
трет-бутанол - пропионовая кислота - вода, 50:12:38 (по объему).
Соскабливают разделившиеся окрашенные зоны с пластинки в различные центрифужные пробирки. Доводят аммиаком рН растворов до 7. Добавляют по 5 или 10 см3 воды, закрывают пробками и встряхивают в течение 1 мин. Удаляют пробки и центрифугируют при 2000 об/мин в течение 10 мин. Декантируют супернатант в другие центрифужные пробирки и повторяют центрифугирование.
1.2.5. Количественное определение
Определяют поглощение чистых растворов на спектрофотометре при характерных для каждого красителя длинах волн максимума поглощения. Для контроля чистоты красителя и подтверждения структуры красителя снимают спектр образца и сравнивают со спектром стандартного раствора или библиотечным спектром.
1.2.5.1. Расчет содержания основного вещества в красителях
Содержание основного вещества X, определенное спектрофотометрически, рассчитывают по формуле:
![]() ,
где
,
где
D - оптическая плотность исследуемого раствора;
V - объем, см3;
Э - коэффициент светопоглощения эталона настоящей методики;
g - масса, мг.
2. Определение синтетических пищевых красителей методом ВЭЖХ
2.1. Условия ОФ ВЭЖХ
Колонка: Kromasil С18, 5 mм, 250´4,6 мм ID.
Элюент: 10 мМ фосфатный буфер, рН 4,2 - ацетонитрил, 80:20 об. %.
Скорость подачи элюента: 1 см3/мин.
Детектирование: UV-Vis 500 нм (красные), 450 нм (желтые) и 600 нм (синие СПК).
2.2. Условия ион-парной ОФ ВЭЖХ
Колонка: Kromasil С18, 5 mм, 250´4,6 мм ГО
Элюент: 10 мМ тетрабутиламмоний фосфат, 10 мМ фосфатный буфер, рН 4,2 -ацетонитрил, 55:45 об. %.
Скорость подачи элюента: 0,9 см3/мин.
Детектирование: UV-Vis 500 нм (красные), 450 нм (желтые) и 600 нм (синие СПК).
Рис. 26. Хроматограмма модельной смеси СПК в условиях ОФ ВЭЖХ. Детектирование: 500 нм.
Рис. 27. Хроматограммы модельной смеси СПК в условиях ион-парной хроматографии. Детектирование: UV-Vis (а) - 600 нм, (б) - 500 нм, (в) - 450 нм.
2.3. Подготовка стандартных растворов
Стандартные растворы синтетических пищевых красителей готовят путем растворения точных навесок СПК с известной концентрацией основного вещества в мерных колбах дистиллированной водой. Эритрозин (Е 127) растворяют в метаноле. Градуировочные графики строят в координатах S (площадь пика) - С (концентрация СПК), г/л в интервале концентраций 0,002 - 0,01 г/л.
2.4. Пробоподготовка
Соки и сокосодержащие БАД (10 см3) разбавляют дистиллированной водой в колбе объемом 100 см3, пробы фильтруют через мембранный фильтр с диаметром пор 0,2 мкм. Концентраты соков (10 г) разбавляют дистиллированной водой в колбе объемом 250 см3, пробы фильтруют через мембранный фильтр с диаметром пор 0,2 мкм.
Глава 5.
МЕТОДЫ ИССЛЕДОВАНИЙ БЕЗОПАСНОСТИ
I. Методы определения микотоксинов
Для обнаружения, идентификации и количественного определения микотоксинов используют следующие методы:
· двумерную и одномерную ТСХ (для серийных определений);
· ВЭЖХ с УФ-фотометрическим и флуориметрическим детектированием (для серийных и арбитражных анализов).
Специфическое оборудование и материалы
Аппарат для встряхивания проб типа АВУ-6С, ТУ 64-1-2451-78.
Ротационный испаритель с ловушкой, модель ИР-2М (завод «Химлабприбор»), или аналогичный.
Мельница лабораторная электрическая ЭМ-3А, ТУ 46-22-236-79, или аналогичная.
Облучатель настольный ультрафиолетовый, снабженный светофильтром с максимумом пропускания при длине волны 365 нм.
Жидкостный хроматограф с насосом высокого давления с подачей растворителя от 0,1 до 5 см3/мин; колонка и предколонка с силикагелем или с силикагелем, химически связанным с октадецилсиланом (силикагель-С18), или с силикагелем, химически связанным с алкилнитрилом, с размером частиц 5 мкм; длина колонок - 25 см, предколонок - 4,5 см, внутренний диаметр колонок - 0,46 см; ультрафиолетовый детектор с переменной длиной волны; флуориметрический детектор; регистрирующий потенциометр (самописец) или интегратор.
Микрошприцы МШ-10 и МШ-25 для жидкостной хроматографии.
Спектрофотометр СФ-26, СФ-46 или подобный, позволяющий проводить измерения при длине волны 275 - 360 нм, с допустимой абсолютной погрешностью измерений коэффициента пропускания не более 1 %.
Кюветы кварцевые с толщиной поглощающего свет слоя 10 мм.
Шкаф сушильный, обеспечивающий нагрев до 150 °С.
Центрифуга лабораторная, обеспечивающая скорость вращения 4000 об/мин, стаканы центрифужные стеклянные вместимостью 200 см3.
Насос водоструйный лабораторный, ГОСТ 25336.
Стеклянные камеры для ТСХ с притертыми крышками, например стеклянный четырехугольный сосуд размером 195´195´200 мм завода «Дружная горка».
Пластинки для ТСХ «Силуфол» размером 15´15 или 20´20 см (Чехия) или подобные.
Распылитель стеклянный с грушей.
Воронки делительные ВД 2-250 или ВД 2-500, ГОСТ 25336.
Колонки стеклянные хроматографические 230´15 и 300´22 мм.
Силикагель L для колоночной хроматографии с размером частиц 100 - 160 мкм, «Lachema» (Чехия).
Флоризил, 60 - 100 меш, «Merk» (Германия).
Ацетон, ч.д.а., ГОСТ 2603-71.
Ацетонитрил, ч., ТУ 6-09-3543-74.
Бензол, ч.д.а., ГОСТ 5955-75.
Гексан, ч., ТУ 6-09-3375-73.
1,4-Диоксан, ч.д.а., ГОСТ 10455.
Изопропиловый спирт (пропанол-2), ч.д.а., ТУ 6-09-402-76.
Метанол, ч., ГОСТ 6995-77.
Спирт этиловый ректификат, ТУ 6-09-1710-77.
Толуол, ч., ГОСТ 5789-78.
Хлороформ медицинский, ГОСТ 3160-51, или технический, ГОСТ 20015.
Этиловый эфир уксусной кислоты (этилацетат), ч., ГОСТ 22300-76.
Эфир диэтиловый медицинский, ГОСТ 6265-52.
Кислота азотная, ч.д.а., ГОСТ 4461-67.
Кислота лимонная, 1-водная, ч.д.а., ГОСТ 3652-29.
Кислота муравьиная, ч.д.а., ГОСТ 5848-73.
Кислота серная, ч., ГОСТ 4204-77.
Кислота соляная, х.ч., ГОСТ 3118.
Кислота уксусная, ч.д.а., ГОСТ 61-75.
Алюминия оксид нейтральный по Брокману 11 для колоночной хроматографии, «Reanal» (Венгрия), номер по каталогу 01125 или алюминия оксид для хроматографии, ТУ 6-09-3916-75.
Алюминий хлористый, 6-водный, х.ч., ГОСТ 3759-75.
Бензидин, ч.д.а., раствор в муравьиной кислоте концентрацией 5 г/дм3.
Йод, ч., ГОСТ 4159, раствор в этиловом эфире концентрацией 250 г/дм3.
Калий хлористый, х.ч., ГОСТ 4234-77.
Калий железосинеродистый 3-водный, х.ч., ГОСТ 4207, водный раствор концентрацией 150 г/дм3 (раствор Карреза 1).
Цинк уксуснокислый, 2-водный, х.ч., ГОСТ 5823, водный раствор концентрацией 300 г/дм3 (раствор Карреза 2).
Калий марганцовокислый, х.ч., ГОСТ 20490, водный раствор концентрацией 15 г/дм3.
Натрия сульфат безводный, х.ч., ГОСТ 4166-76.
Натрий хлористый, х.ч., ГОСТ 4233-66.
Натрий углекислый кислый (гидрокарбонат), х.ч., ГОСТ 4201-79, водный раствор концентрацией 50 г/дм3.
Прочный синий В-соль (диазоль синий С) для гистологии («Chemapol», Чехия), водный раствор концентрацией 10 г/дм3.
Свинец уксуснокислый, х.ч., ГОСТ 1027-67, водный раствор концентрацией 150 г/дм3.
Серебро азотнокислое, ч., ГОСТ 1277, водный раствор концентрацией 250 г/дм3.
Уголь активированный Р.72.270.3.
Бумажные фильтры обеззоленные марки ФОМ.
Приготовление и хранение стандартных растворов микотоксинов
Чистоту и аутентичность стандартных препаратов микотоксинов определяют с помощью УФ-спектрофотометрии (табл. 47) и хроматографических методов (ТСХ, ВЭЖХ).
Расчет концентрации стандартных растворов проводят по данным УФ-спектрофотометрии. Данные о коэффициентах молярной экстинкции микотоксинов в максимумах поглощения в УФ-спектре приведены в табл. 47.
Приготовление и расчет концентрации стандартных растворов микотоксинов
Для приготовления стандартных растворов микотоксинов рекомендуется использовать растворители, очищенные перегонкой. Данные о растворителях и концентрациях стандартных и рабочих растворов микотоксинов приведены в табл. 48.
В мерную колбу вместимостью 100 или 250 см3 помещают навеску микотоксина (1 - 10 мг), взятую с точностью до 0,01 мг, приливают нужное количество ацетонитрила согласно таблице 33 или 20 - 30 см3 бензола, тщательно перемешивают до полного растворения вещества, доводят бензолом до метки и измеряют оптическую плотность по данным УФ-спектрофотометрии против соответствующего растворителя.
При необходимости измерения концентрации стандартного раствора в метаноле 3 см3 бензольного или бензол-ацетонитрильного раствора переносят в чистую пробирку, растворитель упаривают в токе азота досуха, к остатку добавляют 3 см3 метанола и измеряют оптическую плотность полученного метанольного раствора.
Таблица 47
Коэффициенты молярной экстинции микотоксинов
|
Молекулярная масса |
Длина волны максимума поглощения, нм |
Коэффициент молярной экстинкции Е, см2/моль |
||
|
Растворитель |
||||
|
метанол |
бензол-ацетонитрил |
|||
|
1 |
2 |
3 |
4 |
5 |
|
Афлатоксины: |
|
|
|
|
|
В1 |
312,3 |
223 |
22100 |
|
|
265 |
12400 |
|
||
|
3621 |
21800 |
198002 |
||
|
В2 |
314,3 |
223 |
18600 |
|
|
265 |
12100 |
|
||
|
3621 |
24000 |
209002 |
||
|
G1 |
328,3 |
242 |
9600 |
|
|
265 |
9600 |
|
||
|
362 |
17700 |
171002 |
||
|
G2 |
330,3 |
242 |
10500 |
|
|
265 |
9000 |
|
||
|
3621 |
19300 |
182002 |
||
|
Ml |
328,3 |
226 |
23100 |
|
|
265 |
11600 |
|
||
|
3571 |
19000 |
188152 |
||
|
Стеригматоцистин |
324,1 |
3261 |
15310 |
152002 |
|
- |
277 |
3040 |
|
|
|
|
246 |
32870 |
|
|
|
Дезоксиниваленол |
296,0 |
2181 |
45002 |
|
|
Зеараленон |
318,1 |
236 |
28850 |
|
|
274 |
127102 |
|
||
|
316 |
5840 |
|
||
|
Охратоксин А |
403,1 |
214 |
37200 |
|
|
333 |
6400 |
55503 |
||
|
Патулин |
154,0 |
276 |
145002 |
|
1 Длины волн, используемые для определения концентрации стандартных растворов.
2 Растворитель, используемый для определения концентрации стандартного раствора.
3 Для охратоксина А используется смесь бензол - уксусная кислота (99:1).
Рассчитывают концентрацию микотоксина в стандартных растворах по формуле:
![]() ,
где
,
где
С - концентрация исследуемого раствора, мкг/см3 или нг/мкл;
D - оптическая плотность раствора;
М - молекулярная масса микотоксина (табл. 47);
Е - коэффициент молярной экстинкции (табл. 47);
L - толщина слоя раствора, см.
Рабочие растворы стандартов готовят путем разбавления соответствующих стандартных растворов, для чего необходимую аликвоту рабочего раствора микотоксина доводят до соответствующего объема бензолом или смесью бензол - ацетонитрил согласно табл. 48. Например, для приготовления рабочего раствора смеси афлатоксинов b1, В2, g1 и G2 с концентрациями 1,0, 0,5, 0,4 и 0,2 мкг/см3 соответственно в мерную колбу 50 см3 помещают 5,0 см3 стандартного раствора афлатоксина b1, 2,5 см3 раствора В2, 2,0 см3 раствора g1 и 1,0 см3 раствора G2, затем доводят до метки смесью бензол - ацетонитрил (98:2). Для приготовления стандартного раствора афлатоксина m1 с концентрацией 0,2 мкг/см3 в мерную колбу на 50 см3 помещают 1 см3 стандартного раствора афлатоксина m1 и доводят до метки смесью бензол - ацетонитрил (9:1).
Таблица 48
Стандартные и рабочие растворы микотоксинов
|
Растворители для приготовления стандартного раствора |
Концентрации микотоксинов в растворах, мкг/см3 |
||
|
стандартном |
рабочем |
||
|
Афлатоксины: |
|
|
|
|
в1 |
Бензол-ацетонитрил (98:2) |
10 |
1,0 |
|
В2 |
|
10 |
0,5 |
|
G1 |
|
10 |
0,4 |
|
G2 |
|
10 |
0,2 |
|
М1 |
Бензол-ацетонитрил (9:1) |
10 |
0,2 |
|
Стеригматоцистин |
Бензол |
10 |
10,0 |
|
Дезоксиниваленол |
Бензол-ацетонитрил (5:1) |
25 |
10,0 |
|
Зеараленон |
Бензол |
100 |
10,0 |
|
Охратоксин А |
Бензол-уксусная кислота (99:1) |
50 |
5,0 |
|
Патулин |
Бензол-ацетонитрил (9:1) |
10 |
10,0 |
Если используют расфасованные во флаконы кристаллические стандарты микотоксинов, полученные от фирм-поставщиков, поступают следующим образом: во флакон с веществом добавляют, согласно табл. 48, необходимое количество растворителя и тщательно перемешивают до полного растворения микотоксинов, содержимое флакона количественно переносят в мерную колбу с объемом, необходимым для получения концентрации стандартного раствора микотоксина, указанной в соответствующей колонке табл. 48. Флакон тщательно промывают несколькими порциями растворителя, каждый раз перенося содержимое флакона в мерную колбу, затем доводят объем до метки растворителем. Измеряют концентрацию полученного раствора по данным УФ-спектрофотометра, как описано выше.
Хранение стандартных растворов микотоксинов
Стандартные растворы микотоксинов следует хранить в стеклянной посуде с притертыми пробками в темном прохладном месте (при температуре около 0 °С) до одного года и использовать для приготовления рабочих стандартных растворов.
Перед употреблением в работе стандартные растворы следует довести до комнатной температуры и только после этого открывать пробки.
В процессе хранения стандартных растворов вследствие испарения растворителей могут происходить некоторые изменения их концентраций. По этой причине следует проводить их систематический контроль (1 - 2 раза в квартал) как описано выше.
1. Метод обнаружения, идентификации и определения содержания афлатоксинов в БАД на зерновой и зернобобовой основе с помощью тонкослойной и высокоэффективной жидкостной хроматографии
Экстракция
Отобранную пробу измельчают в течение 1 - 2 мин в кофемолке или лабораторной мельнице. Навеску 25 г измельченного БАД помещают в плоскодонную коническую колбу на 250 см3, тщательно перемешивают с 25 см3 10 % раствора хлорида натрия. Добавляют 0,0125 см3 стандартного раствора афлатоксина B2 (внутренний стандарт), что соответствует загрязнению пробы афлатоксином на уровне 1 ПДК, перемешивают. Добавляют 100 см3 ацетона и встряхивают на аппарате для встряхивания в течение 30 мин. Полученную смесь фильтруют через бумажный складчатый фильтр, отбирают 50 см3 фильтрата.
Очистка экстракта
К 50 см3 фильтрата добавляют 20 см3 15 % раствора ацетата свинца и 30 см3 дистиллированной воды, перемешивают и оставляют на 10 мин в темноте. Отфильтровывают образовавшийся осадок через бумажный складчатый фильтр, отбирают 80 см3 фильтрата. Встряхивают в делительной воронке с гексаном (2´30 см3), каждый раз отбрасывая верхний гексановый слой. Водно-ацетоновый слой экстрагируют хлороформом (1 раз 40 см3, 2 раза 30 см3 смеси хлороформ-ацетон 3:1). Объединенные хлороформные экстракты помещают в плоскодонную колбу на 250 см3, для удаления воды добавляют 5 - 7 г безводного сульфата натрия, встряхивают и оставляют на 30 мин в темноте. Высушенный раствор фильтруют через химическую воронку с комочком ваты в грушевидную колбу, сульфат натрия промывают 10 см3 хлороформа, добавляя смыв к фильтрату. Хлороформный экстракт упаривают на ротационном испарителе досуха. Остаток растворяют в 0,4 см3 смеси бензол - ацетонитрил (98:2) - раствор А и анализируют с помощью ТСХ.
Обнаружение афлатоксинов с помощью одномерной ТСХ
Пластинку для ТСХ («Силуфол») размечают тонкими карандашными линиями в соответствии с рис. 28. На горизонтальной линии, проведенной в 1,5 см от нижнего края пластинки, наносят с помощью микрошприца в 2 см друг от друга по 0,002, 0,005 и 0,01 см3 рабочих растворов афлатоксина b1 (2, 5, и 10 нг соответственно) и афлатоксина В2 (1, 2,5 и 5 нг соответственно) или рабочего раствора смеси афлатоксинов b1, B2, g1 и G2. На расстоянии 1 см между пятнами стандартов наносят 0,01 и 0,02 см3 раствора А. Пластинку помещают в камеру для ТСХ со смесью эфир - метанол - вода (94:4,5:1,5) и развивают пластинку до достижения фронтом растворителя линии, проведенной в 1 см от верхнего края пластинки. Пластинку извлекают из камеры, сушат на воздухе 5 мин и рассматривают в длинноволновом УФ-свете. Обнаружение на пластинке пятен, соответствующих по хроматографической подвижности и цвету флуоресценции пятнам стандартов афлатоксинов, свидетельствует о возможном наличии афлатоксинов в БАД. Наличие пятна внутреннего стандарта свидетельствует о правильном извлечении афлатоксинов из пробы.
Тесты, подтверждающие наличие афлатоксинов в БАД
Тест первый
Для подтверждения наличия афлатоксинов ТСХ-пластинку опрыскивают раствором азотной кислоты в воде (1:2) и рассматривают ее в длинноволновом УФ-свете. Если цвет флуоресценции стандартов афлатоксинов изменился с синего (В1, B2) или сине-зеленого (G1, G2) на желтый, а цвет флуоресценции пятен экстракта на желтый не изменился, то афлатоксины в пробе отсутствуют. Если же цвет флуоресценции пятен экстракта также изменился на желтый, то это служит подтверждением возможного наличия афлатоксинов в БАД.
Тест второй
На стеклянную пластинку 20´20 см наносят 2 - 4 см3 5 - 10 %-ного раствора йода в эфире, распределяют его по всей поверхности и дают испариться. Над ТСХ-пластинкой на расстоянии 0,5 - 1 см помещают пластинку с тонким слоем йода и подвергают ее воздействию паров йода в течение 1 - 2 мин. Затем пластинку рассматривают в длинноволновом УФ-свете. Сохранение цвета и интенсивности флуоресценции пятен стандартов и соответствующих им пятен экстракта подтверждает возможное наличие афлатоксинов в БАД.
Подтверждение наличия афлатоксинов и количественное определение афлатоксина b1 с помощью двумерной ТСХ
Пластинку «Силуфол» размечают тонкими карандашными линиями согласно рис. 29. В правом нижнем углу на расстоянии 1,5 см от краев наносят 0,02 см3 раствора А, в правом верхнем углу наносят 0,005 см3 рабочего раствора смеси афлатоксинов или рабочих растворов афлатоксинов b1и B2. В левом нижнем углу на расстоянии 1, 2 и 3 см от левого края и 1,5 см от нижнего края пластинки наносят 0,002, 0,004 и 0,006 см3 рабочего раствора смеси афлатоксинов или рабочих растворов афлатоксинов b1и B2. Пластинку помещают в камеру для ТСХ со смесью эфир - метанол - вода (94:4,5:1,5) и развивают пластинку в первом направлении до достижения фронтом растворителя тонкой карандашной линии, проведенной в 3 см от верхнего края пластинки. Пластинку извлекают и сушат на воздухе 5 мин, затем развитие пластинки проводят во втором направлении в системе хлороформ - ацетон - вода (90:10:1). После достижения фронтом растворителя карандашной линии ее извлекают, сушат на воздухе и рассматривают в длинноволновом УФ-свете. Обнаружение афлатоксинов в растворе А производят аналогично описанному выше. В случае подтверждения наличия афлатоксинов сравнивают интенсивность флуоресценции пятна стандарта b1 с пятном афлатоксина b1 в растворе А и определяют количество вещества в пятне. Расчет содержания афлатоксина b1 в образце производят по формуле:
![]() ,
где
,
где
С - концентрация афлатоксина b1 в образце, мг/кг;
![]() - объем
водно-ацетоновой смеси для экстракции, см3 (125 см3);
- объем
водно-ацетоновой смеси для экстракции, см3 (125 см3);
V2 - объем водно-ацетонового фильтрата, взятый для анализа, см3 (50 см3);
V3 - объем водно-ацетонового фильтрата и раствора ацетата свинца, см3 (100 см3);
V4 - объем водно-ацетонового фильтрата после очистки ацетатом свинца, см3 (80 см3);
V5 - объем экстракта перед ТСХ (раствор А), см3 (0,4 см3);
V6 - объем экстракта (раствор А), наносимый на пластинку, см3 (0,02 см3);
М - навеска образца, взятая для анализа, г (25 г);
т - масса афлатоксина b1 в V6 см3 экстракта, оцененная по ТСХ, нг;
К - степень извлечения афлатоксина В1 по табл. 50.
При приведенных в скобках объемах и навесках формула содержания афлатоксина В1 выражается следующим образом:
![]() ,
мг/кг
,
мг/кг
Если интенсивность флуоресценции афлатоксина В1 в экстракте выше интенсивности флуоресценции пятна стандарта, соответствующего 0,006 см3 рабочего раствора (6 нг афлатоксина В1), на пластинку следует нанести либо меньшее количество экстракта (уменьшить объем V6), либо разбавить раствор А смесью бензол - ацетонитрил (98:2) (увеличить объем V5), внеся соответствующие коррективы в расчетную формулу.
По интенсивности флуоресценции внутреннего стандарта можно оценить степень извлечения афлатоксинов из пробы.
Очистка экстракта перед ВЭЖХ
В случае анализа экстракта на содержание афлатоксинов методом ВЭЖХ необходимо произвести его очистку с помощью колоночной хроматографии. На дно стеклянной колонки размером 230´5 мм помещают кусочек ваты, присыпают безводный сульфат натрия (толщина слоя 5 мм), заливают суспензию 2 г силикагеля в бензоле и, не давая бензолу стечь (колонка закрыта снизу), насыпают слой безводного сульфата натрия (20 мм). Дают бензолу стечь и на колонку наносят экстракт образца (раствор А). Грушевидную колбу из-под экстракта ополаскивают смесью бензол - ацетонитрил (98:2) 2 раза по 0,3 см3 и раствор наносят на колонку, бензольный элюат отбрасывают. Колонку элюируют 60 см3 смеси хлороформ - ацетон (9:1), элюат фильтруют через бумажный или капроновый фильтр, упаривают досуха на ротационном испарителе, остаток растворяют в 0,4 см3 смеси бензол - ацетонитрил (98:2) (очищенный раствор А) и анализируют с помощью ВЭЖХ.
Обнаружение и количественное определение афлатоксинов b1, b2, G1 и G2 с помощью ВЭЖХ
Условия и параметры хроматографического анализа приведены в табл. 49.
Таблица 49
Условия хроматографического анализа афлатоксинов
|
Подвижная фаза |
Коэффициент емкости, К’ для |
|||||
|
B1 |
В2 |
G1 |
G2 |
М1 |
||
|
Силикагель |
Эфир - метанол - вода, 95:4:1 |
1,1 - 1,5 |
1,8 - 2,1 |
2,2 - 2,5 |
3,3 - 3,6 |
2,6 - 2,9 |
|
Силикагель |
Эфир - метанол - вода, 90:8:2 |
0,9 - 1,3 |
1,2 - 1,5 |
1,4 - 1,7 |
1,8 - 2,1 |
1,4 - 1,7 |
|
Силикагель |
Толуол - этилацетат - 85 % муравьиная кислота, 80:40:9,5 |
1,2 - 1,5 |
1,8 - 2,1 |
2,0 - 2,3 |
3,1 - 3,4 |
3,0 - 3,3 |
Время удерживания токсина на колонке при конкретных условиях анализа целесообразно выражать через коэффициент ее емкости, средние значения которого приведены в данной табл. 49. Коэффициент емкости характеризует удерживание анализируемого компонента (афлатоксина) в единицах мертвого объема колонки и определяется по формуле:
![]() ,
где
,
где
К¢ - коэффициент емкости данной колонки;
Т0 - мертвый объем системы, соответствующий времени выхода растворителя;
Т1 - время удерживания афлатоксина.
Рекомендуется использовать перегнанные метанол, толуол, этилацетат и дистиллированную воду. Эфир перед использованием пропускают через колонку с оксидом алюминия. Все растворители необходимо предварительно отфильтровать.
Для достижения необходимого предела обнаружения при ВЭЖХ афлатоксинов групп В, G и М с использованием подвижной фазы эфир - метанол - вода необходимо заполнить кювету флуориметрического детектора силикагелем типа «Силасорб 600» с размером частиц 5 - 8 мкм.
Флуориметрический детектор устанавливают на длину волны возбуждающего излучения 360 нм (или устанавливают соответствующий фильтр на линии возбуждения при работе с флуориметрическим детектором без монохроматора), на линии эмиссии устанавливают эмиссионный фильтр с полосой пропускания от 400 или 420 нм. Входное напряжение самописца - 10 мВ, чувствительность детектора устанавливают таким образом, чтобы 1 - 2 нг афлатоксина b1 соответствовало отклонению пера на полную шкалу самописца при уровне шума от 3 до 5 % от полной шкалы.
Для калибровки прибора по флуориметрическому детектору в инжектор с помощью микрошприца вводят 0,001 см3 рабочего раствора смеси стандартов для ВЭЖХ или по 0,001 см3 рабочих растворов афлатоксинов b1 и B2 (соответствует 1,0 нг афлатоксина b1, 0,5 нг B2, 0,4 нг g1 и 0,2 нг G2) и затем 0,002 см3 рабочего раствора (2,0 нг b1, 1,0 нг B2, 0,8 нг g1 и 0,4 нг G2). Для каждого количества афлатоксинов определяют высоту пика. Рассчитывают калибровочные коэффициенты для афлатоксинов по следующей формуле:
![]() ,
где
,
где
K - калибровочный коэффициент для афлатоксинов b1, g1 или G2;
С - концентрация афлатоксинов b1, g1 или G2 в рабочих растворах, мкг/см3;
С1 - концентрация афлатоксина В2 (внутренний стандарт) в рабочем растворе, мкг/см3;
h1 - высота пика афлатоксина В2 (внутренний стандарт), мм;
h - высота пика афлатоксина b1, g1 или G2, мм.
При высоких уровнях загрязнения БАД афлатоксинами (свыше 0,1 мг/кг по афлатоксину b1) возможно использование также ВЭЖХ с УФ-детектором. Калибровку прибора по УФ-детектору проводят так же, как в случае флуориметрического детектора, вводя в петлю инжектора 0,010, 0,015 и 0,020 см3 рабочего раствора (соответствует 10, 15 и 20 нг афлатоксина b1; 5, 7,5 и 10 нг афлатоксина В2; 4, 6 и 8 нг G1; 2, 3 и 4 нг G2).
В инжектор хроматографа вводят с помощью микрошприца 0,02 см3 раствора А (20 мкл). При наличии пика, совпадающего по времени удерживания с афлатоксином b1 (B2, g1, G2); определяют его высоту (h).
Расчет концентрации афлатоксинов проводят по формуле:
![]() ,
где
,
где
С - концентрация афлатоксина в БАД, мг/кг;
K - калибровочный коэффициент для афлатоксинов b1 (или g1, G2);
h - высота пика афлатоксинов b1 (или g1, G2);
h1 - высота пика афлатоксина В2 (внутренний стандарт), внесенный в пробу, см3;
Сст - концентрация афлатоксина В2 (внутренний стандарт) в стандартном растворе, мкг/см3, (см. табл. 48);
V - объем стандартного раствора афлатоксина В2 (внутренний стандарт), внесенный в пробу, см3;
т - навеска образца, взятая для анализа, г.
Если пик афлатоксина выходит за пределы шкалы самописца, анализ с помощью ВЭЖХ проводят повторно, уменьшая объем вводимого в инжектор раствора экстракта или разбавляя раствор очищенного экстракта смесью бензол - ацетонитрил (98:2). При необходимости дополнительного подтверждения проводят совместную ВЭЖХ экстракта со стандартом афлатоксинов.
Таблица 50
Относительные допустимые внутрилабораторные (Rr) и межлабораторные (RR) расхождения результатов определения, величина предела определения и степень извлечения для методов определения микотоксинов В1
|
Уровень загрязнения, г/кг |
Метод |
Rsr, % |
RSR, % |
Rr, % |
RR, % |
Предел определения, мг/кг |
Степень извлечения, % |
|
|
|
0,02 |
ТСХ |
26 |
50 |
73 |
140 |
0,001 |
80 |
|
|
|
ВЭЖХ |
7 |
14 |
20 |
40 |
0,00015 |
70 |
|
|
0,003 |
ТСХ |
38 |
59 |
106 |
166 |
0,001 |
75 |
|
|
|
ВЭЖХ |
13 |
23 |
36 |
64 |
0,00015 |
80 |
Метрологические характеристики
Относительное допустимое расхождение между результатами двух параллельных определений, выполненных в одной лаборатории, по отношению к среднему арифметическому значению (Rr) и относительное допустимое расхождение между результатами испытаний, выполненных в двух разных лабораториях, по отношению к среднему арифметическому значению (RR), а также предел определения по данным авторов методов анализа микотоксинов В1 и m1 приведены в табл. 50, в которой приведены также относительные внутрилабораторные (Rsr) и межлабораторные (RSR) среднеквадратичные отклонения и средняя степень извлечения микотоксинов.
2. Метод обнаружения, идентификации и определения содержания охратоксина А
Экстракция
Навеску 25 г отобранной измельченной пробы помещают в плоскодонную коническую колбу на 250 см3, добавляют 12,5 см3 1 % раствора уксусной кислоты в воде и 125 см3 хлороформа. Встряхивают на аппарате для встряхивания в течение 30 мин. Полученную смесь фильтруют через бумажный складчатый фильтр в мерный цилиндр вместимостью 100 см3, отбирают 50 см3 фильтрата.
Очистка экстракта
В делительную воронку переносят 50 см3 фильтрата, добавляют 35 см3 3 % раствора гидрокарбоната натрия в смеси метанол - вода (1:4). Встряхивают, после разделения слоев верхний водный слой отделяют. Нижний хлороформный слой экстрагируют (2 раза по 35 см3) водно-метанольным раствором гидрокарбоната натрия. Объединенные водные экстракты встряхивают в делительной воронке с хлороформом (2´25 см3), каждый раз после разделения слоев отбрасывая нижний хлороформный слой. Водный экстракт подкисляют раствором серной кислоты в воде с концентрацией 2 моль/дм3 до рН 2 - 3 по универсальной индикаторной бумаге (примерно 9,5 - 10 см3 раствора серной кислоты). Подкисленный водный раствор немедленно экстрагируют в делительной воронке хлороформом (1 раз по 50 см и 2 раза по 25 см3). Объединенные хлороформные экстракты сушат безводным сульфатом натрия (10 - 12 г) в течение 0,5 ч. Раствор порциями фильтруют через химическую воронку с кусочком ваты в грушевидную колбу, осушитель промывают 15 см3 хлороформа, который также фильтруют в ту же колбу, и раствор упаривают досуха на ротационном испарителе при температуре водяной бани не выше 40 - 45 °С. Остаток растворяют в 0,4 см3 смеси бензол - уксусная кислота (99:1) для проведения ТСХ анализа или в 0,4 см3 метанола для проведения анализа с помощью ВЭЖХ (раствор А).
Обнаружение и количественное определение охратоксина А с помощью двумерной ТСХ
Пластинку «Силуфол» размечают тонкими карандашными линиями, не повреждая слой силикагеля, согласно рис. 29. В правом нижнем углу на расстоянии 1,5 см от краев пластинки наносят с помощью микрошприца 0,02 см3 раствора А, в правом верхнем углу наносят 0,002 и 0,004 см3 рабочего раствора охратоксина А (10 и 20 нг соответственно). В левом нижнем углу пластинки наносят 0,001; 0,003 и 0,005 см3 рабочего раствора охратоксина А (5; 15 и 25 нг охратоксина А соответственно).
Пластинку помещают в хроматографическую камеру для ТСХ со смесью эфир - гексан - хлороформ - муравьиная кислота (30:30:30:1) и элюируют в первом направлении до достижения фронтом растворителя тонкой карандашной линии, проведенной в 3 см от верхнего края пластинки.
Пластинку извлекают из камеры и сушат на воздухе 5 мин. Затем проводят элюирование во втором направлении в системе толуол - этилацетат - муравьиная кислота (55:35:10). После достижения фронтом растворителя тонкой карандашной линии, проведенной в 3,5 см от верхнего края пластинки, ее извлекают из камеры и сушат на воздухе.
Затем пластинку рассматривают в длинноволновом УФ-свете. В этих условиях пятна охратоксина А флуоресцируют зелено-синим цветом. Пластинку опрыскивают насыщенным раствором карбоната натрия в воде и рассматривают в длинноволновом УФ-свете. Цвет флуоресценции охратоксина А должен измениться на синий. Обнаружение на пластинке пятна, соответствующего по цветам флуоресценции и хроматографической подвижности в двух системах растворителей пятнам стандарта охратоксина А, свидетельствует о наличии охратоксина А в образце. Сравнивая интенсивность флуоресценции разных количеств стандарта охратоксина А с интенсивностью флуоресценции пятна охратоксина А в образце, оценивают количество нг охратоксина А в нанесенном на пластинку объеме раствора А.
Концентрацию охратоксина А в образце рассчитывают по формуле:
![]() ,
где
,
где
С - концентрация охратоксина А в образце, мг/кг;
V1 - объем хлороформа для экстракции образца, см3 (125 см3);
V2 - объем хлороформного фильтрата, взятый для анализа, см3 (50 см3);
V3 - объем экстракта для ТСХ (раствор А), см3 (0,4 см3);
V4 - объем экстракта (раствора А), нанесенный на пластинку, см3 (0,02 см3);
т - количество охратоксина А в пятне, оцененное по ТСХ, нг;
М - навеска образца, взятая для анализа, г (25 г);
K - степень извлечения охратоксина А по табл. 51.
При приведенных в скобках объемах и навесках формула концентрации охратоксина А в образце выражается следующим образом:
![]() ,
мг/кг.
,
мг/кг.
Если интенсивность флуоресценции пятна охратоксина А в экстракте выше интенсивности флуоресценции пятна стандарта, соответствующего 0,005 см3 стандартного раствора (25 нг охратоксина А), то на пластинку следует нанести либо меньшее количество экстракта (уменьшить объем V4), либо разбавить раствор А смесью бензол - уксусная кислота 99:1 (увеличить объем V3), повторно провести ТСХ и внести соответствующие коррективы в расчетную формулу.
Обнаружение и количественное определение охратоксина А с помощью ВЭЖХ
Приготовление стандартного раствора охратоксина А для ВЭЖХ
0,2 см3 стандартного раствора охратоксина А концентрацией 50 мкг/см3 помещают в мерную колбу на 10 см3, растворитель удаляют в токе азота, остаток растворяют в метаноле и доводят метанолом до метки. Получают стандартный раствор охратоксина А для ВЭЖХ концентрацией 1 мкг/см3 (1 нг/мкл).
Количественное определение охратоксина А с помощью обращеннофазной ВЭЖХ
Условия ВЭЖХ: подвижная фаза метанол - вода - уксусная кислота (75:25:1,5), расход подвижной фазы - 1 см3/мин. Рекомендуется использовать перегнанный метанол и бидистиллированную воду, фильтруя их через бумажный складчатый фильтр перед использованием. Флуориметрический детектор устанавливают на длину волны возбуждающего излучения 330 - 333 нм (или устанавливают соответствующий фильтр на линии возбуждения при работе с флуориметрическим детектором без монохроматора), на линии эмиссии устанавливают эмиссионный фильтр с границей пропускания от 415 - 420 нм. Входное напряжение самописца 10 мВ, чувствительность детектора устанавливают таким образом, чтобы 5 нг охратоксина А соответствовало отклонение пера на полную шкалу самописца при уровне шума от 3 до 5 % от полной шкалы.
Для калибровки прибора в инжектор с помощью микрошприца вводят 0,002; 0,003 и 0,005 см3 (2; 3 и 5 нг) рабочего раствора охратоксина А для ВЭЖХ. Для каждого количества введенного охратоксина А определяют высоту пика. При описанных выше условиях коэффициент емкости (К¢) для охратоксина А составляет примерно 1,9 - 2,5. В инжектор хроматографа вводят с помощью микрошприца 0,02 см3 раствора охратоксина А в метаноле. При наличии пика, совпадающего по времени удерживания с охратоксином А, определяют его высоту (h).
Расчет концентрации охратоксина А в образце производят по формуле:
![]() ,
где
,
где
С - концентрация охратоксина А в образце, мг/кг;
V1 - объем хлороформа для экстракции образца, см3 (125 см3);
V2 - объем хлороформного фильтрата, взятый для анализа, см3 (50 см3);
V3 - объем экстракта (раствор А) перед ВЭЖХ, см3 (0,4 см3);
V4 - объем экстракта (раствор А), введенный в хроматограф, см3 (0,02 см3);
т - масса стандарта охратоксина А, введенного в хроматограф, нг;
hcm - высота пика, соответствующая данной массе стандарта, мм;
h - высота пика охратоксина из образца, мм;
М - навеска образца для анализа, г (25 г);
k - степень извлечения охратоксина А по табл. 51.
Если пик охратоксина А в образце выходит за пределы шкалы самописца, анализ с помощью ВЭЖХ проводят повторно после разбавления экстракта (раствор А) метанолом (после увеличения объема V3).
Метрологические характеристики
Относительное допустимое расхождение между результатами двух параллельных определений, выполненных в одной лаборатории, по отношению к среднему арифметическому значению (Rr) и относительное допустимое расхождение между результатами испытаний, выполненных в двух разных лабораториях, по отношению к среднему арифметическому значению (RR), а также предел определения по данным авторов методов анализа охратоксина А приведены в табл. 51, в которой приведены также относительные внутрилабораторные (Rsr) и межлабораторные (RSR) среднеквадратичные отклонения и средняя степень извлечения микотоксинов.
Таблица 51
Относительные допустимые внутрилабораторные (Rr) и межлабораторные (RR) расхождения результатов определения, величина предела определения и степень извлечения для методов определения охратоксина А
|
Метод |
Rsr, % |
RSR, % |
Rr, % |
RR, % |
Предел определения, мг/кг |
Степень извлечения, % |
|
|
0,1 |
тСх |
26 |
43 |
73 |
120 |
0,015 |
90 |
|
|
вэжх |
9 |
19 |
25 |
53 |
0,0013 |
90 |
|
0,03 |
тСх |
34 |
50 |
95 |
140 |
0,015 |
90 |
|
|
вэжх |
19 |
24 |
53 |
67 |
0,0013 |
90 |
3. Метод обнаружения, идентификации и определения содержания дезоксиниваленола (вомитоксина) и зеараленона в БАД на зерновой основе
Экстракция
Навеску 25 г измельченной отобранной пробы помещают в плоскодонную коническую колбу на 250 см3, добавляют 125 см3 смеси ацетонитрил - вода (84:16). Встряхивают на аппарате для встряхивания проб в течение 30 мин. Полученную смесь фильтруют через бумажный складчатый фильтр в мерный цилиндр. Отбирают 25 см3 фильтрата для анализа на дезоксиниваленол и 50 см3 фильтрата для анализа на зеараленон.
Обнаружение, идентификация и определение содержания дезоксиниваленола в экстракте
Очистка экстракта
В стеклянную хроматографическую колонку на дно помещают кусочек ваты, насыпают 0,75 г порошка активированного угля и сверху - слой 0,75 г оксида алюминия.
Над слоем оксида алюминия помещают кусочек ваты. Осторожно наливают в колонку 25 см3 экстракта, соответствующие 5 г исходного образца. Отбирают элюат и, не давая колонке просохнуть, добавляют 10 см3 смеси ацетонитрил - вода (84:16).
Объединенные элюаты фильтруют через бумажный складчатый фильтр в грушевидную колбу на 50 см3, бумажный фильтр промывают 5 - 10 см3 изопропилового спирта в ту же колбу и фильтрат упаривают на ротационном испарителе до объема 5 - 7 см3. Добавляют около 20 см3 изопропилового спирта и повторно упаривают на ротационном испарителе досуха. Остаток в колбе после упаривания не должен содержать капель воды. Остаток растворяют в 0,2 см3 смеси бензол - ацетонитрил (5:1) и плотно закрывают стеклянной пробкой (раствор А). Для обращеннофазной ВЭЖХ остаток растворяют в 0,2 см3 ацетонитрила (раствор a1).
Обнаружение и количественное определение дезоксиниваленола с помощью одномерной ТСХ
Пластинку «Силуфол» размечают в соответствии с рис. 28. На линию, проведенную в 1,5 см от нижнего края пластинки, с помощью микрошприца наносят 0,002; 0,005; 0,01 и 0,02 см3 раствора А. Между пятнами экстракта на расстоянии 1 см от них на ту же линию наносят 0,002; 0,004; 0,006 см3 стандартного раствора дезоксиниваленола (50; 100 и 150 нг дезоксиниваленола). Пластинку помещают в камеру для ТСХ и элюируют в системе гексан - ацетон (3:2) на расстояние 15 см. Пластинку извлекают из камеры, сушат на воздухе 3 - 4 мин и опрыскивают 10 % раствором хлористого алюминия в этаноле. Пластинку нагревают в сушильном шкафу в течение 5 - 7 мин. при 105 °С, затем рассматривают в длинноволновом УФ-свете. Дезоксиниваленол проявляется в виде пятен с синей флуоресценцией с Rf 0,25 - 0,30. Наличие в экстракте пятен, соответствующих по цвету флуоресценции и хроматографической подвижности стандарту дезоксиниваленола, свидетельствует о возможном наличии этого токсина в образце. Для количественного определения сравнивают интенсивность флуоресценции разных количеств стандартов дезоксиниваленола с интенсивностью флуоресценции их пятен в образце, визуально оценивая количество нг токсинов в нанесенных на пластинку объемах раствора А. Концентрацию дезоксиниваленола в образце рассчитывают по формуле:
![]() ,
мг/кг, где
,
мг/кг, где
V1 - объем раствора А, см (0,2 см3);
V2 - объем раствора А, нанесенный на пластинку, см3;
m - масса дезоксиниваленола в V2 см3 раствора А, оцененная визуальным сравнением со стандартом на ТСХ-пластинке, нг;
М - аликвотная навеска образца, соответствующая раствору А (5 г для пшеницы, 3 г для кукурузы);
K - степень извлечения дезоксиниваленола по табл. 54.
Если интенсивность флуоресценции пятна дезоксиниваленола в экстракте выше интенсивности флуоресценции пятна дезоксиниваленола соответствующего 0,006 см3 стандартного раствора, то следует разбавить раствор А смесью бензол - ацетонитрил (5:1), т.е. увеличить объем V1, внеся соответствующие коррективы в расчетную формулу. Окончательное заключение о наличии и уровне загрязнения образца дезоксини-валенолом принимается только на основе данных двумерной ТСХ.
Подтверждение наличия и количественное определение дезоксиниваленола с помощью двумерной ТСХ
Пластинку «Силуфол» размечают тонкими карандашными линиями, не повреждая слоя силикагеля, согласно рис. 30. В правом нижнем углу на расстоянии 1,5 см от краев пластинки наносят с помощью микрошприца 0,02 см3 раствора А. В левом нижнем углу пластинки наносят 0,002; 0,004 и 0,006 см3 стандартного раствора дезоксиниваленола, 0,002 и 0,005 см3 раствора А. В верхнем правом углу пластинки наносят 0,002; 0,004 и 0,006 см3 стандартного раствора дезоксиниваленола, 0,01 и 0,02 см3 раствора А. Пластинку помещают в камеру для ТСХ со смесью гексан - ацетон (3:2) и элюируют ее в первом направлении до достижения фронтом растворителя тонкой карандашной линии, проведенной в 5,5 см от верхнего края, пластинку извлекают из камеры и сушат на воздухе. Затем проводят элюирование пластинки во втором направлении смесью хлороформ - ацетон - изопропиловый спирт (78:12:10). Для элюирования пластинки во втором направлении можно также использовать смесь: эфир - гексан - изопропиловый спирт - вода (77:18:4,5:0,5). После достижения фронтом растворителя карандашной линии, проведенной в 5,5 см от верхнего края пластинки, ее извлекают из камеры и сушат на воздухе.
Обнаружение и количественное определение проводят аналогично описанному выше.
Обнаружение и количественное определение дезоксиниваленола с помощью ВЭЖХ
Условия и параметры анализа дезоксиниваленола с помощью ВЭЖХ приведены в табл. 52.
Расход подвижной фазы 1,0 см3/мин. Рекомендуется использовать перегнанные растворители, фильтруя их через бумажный складчатый фильтр перед использованием. УФ-детектор устанавливают на длину волны 220 - 224 нм, шкала чувствительности 0,01 или 0,005 е.о.п. Для калибровки прибора в инжектор с помощью микрошприца вводят по 0,002 и 0,004 см3 стандартных растворов, что соответствует 50 и 100 нг дезоксиниваленола (при обращеннофазной ВЭЖХ 1 см3 стандартного раствора дезоксиниваленола в смеси бензол - ацетонитрил (5:1), упаривают досуха в токе азота и растворяют в 1 см3 ацетонитрила).
Таблица 52
Условия анализа дезоксиниваленола
|
Сорбент |
Подвижная фаза |
Примерный коэффициент емкости К’ |
|
|
Нормальнофазный |
Силикагель |
Гексан - изопропиловый спирт - вода (75:25:1,5) |
1,3 - 2,0 |
|
Обращеннофазный |
ОДС-силикагель С18 |
Метанол - вода (25:75) |
1,0 - 1,5 |
|
Обращеннофазный |
ОДС-силикагель С18 |
Ацетонитрил - вода (10:90) |
1,5 - 2,0 |
Для каждого количества стандарта определяют высоту пика на хроматограмме. В инжектор хроматографа вводят 10 мкл раствора А (или a1). При наличии пика, совпадающего по времени удерживания со стандартом дезоксиниваленола, определяют его высоту (h). Расчет концентрации дезоксиниваленола в образце проводят по формуле:
![]() ,
где
,
где
С - концентрация дезоксиниваленола в образце, мг/кг;
V1 - объем раствора А, см3 (0,2 см3);
V2 - объем раствора А, внесенный в хроматограф, см3 (0,01 см3);
т - масса стандарта дезоксиниваленола, введенная в хроматограф, нг;
М - аликвотная навеска образца, соответствующая раствору А (5 г);
hсm - высота пика, соответствующая данной массе стандарта, мм;
h - высота пика дезоксиниваленола из образца, мм;
K - степень извлечения дезоксиниваленола - по табл. 54.
Если пик дезоксиниваленола в образце выходит за пределы шкалы самописца, анализ проводят повторно после разбавления раствора A (A1) смесью бензол - ацетонитрил (при нормальнофазной ВЭЖХ) или ацетонитрилом (при обращеннофазной ВЭЖХ), т.е. после увеличения объема V1.
Обнаружение, идентификация и определение содержания зеараленона
Очистка экстракта
В делительную воронку на 500 см3 помещают 50 см3 фильтрата (раздел «Экстракция»), добавляют 50 см3 гексана (или гептана), насыщенного ацетонитрилом. Встряхивают, после разделения слоев отбрасывают верхний гексановый слой. Нижний ацетонитрильный слой дважды встряхивают с 30 см3 гексана, насыщенного ацетонитрилом, каждый раз отбрасывая верхний гексановый слой. К обезжиренному ацетонитрильному экстракту в делительной воронке добавляют 150 см3 дистиллированной воды и 60 см3 бензола. Встряхивают и после разделения слоев отделяют верхний бензольный слой. Если полного расслоения жидкостей не происходит, добавляют 10 см3 насыщенного раствора хлорида натрия и смесь еще раз слегка встряхивают. Водный слой экстрагируют еще дважды 30 см3 бензола, бензольные слои объединяют и сушат безводным сульфатом натрия.
После фильтрования упаривают в грушевидной колбе на 100 см3 на ротационном испарителе при температуре водяной бани не выше 45 °С. Сульфат натрия промывают 10 см3 бензола в ту же грушевидную колбу и упаривают раствор досуха. Остаток растворяют в 0,5 см3 бензола - раствор В.
Обнаружение и идентификация зеараленона с помощью одномерной ТСХ
Пластинку «Силуфол» размечают в соответствии с рис. 28. На линию, проведенную в 1,5 см3 от нижнего края пластинки, с помощью микрошприца, наносят 0,005 и 0,01 см3 раствора В. Между пятнами экстракта на расстоянии 1 см от них на ту же линию наносят 0,005; 0,01 и 0,02 см3 рабочего раствора зеараленона (50; 100 и 200 нг зеараленона, соответственно). Аналогично готовят вторую пластинку «Силуфол». Обе пластинки помещают в камеру для ТСХ и элюируют в системе гексан - ацетон (7:3). Пластинки извлекают, сушат на воздухе 3 - 4 мин. Для обнаружения пятен зеараленона на первой пластинке используют опрыскивание 10 % раствором хлорида алюминия в этиловом спирте с последующим нагреванием в сушильном шкафу при температуре 100 - 105 °С в течение 10 мин. Обнаружение на пластинке пятен, соответствующих по цвету флуоресценции (синий) и хроматографической подвижности пятнам стандартов зеараленона, свидетельствует о возможном наличии зеараленона в образце.
Для подтверждения наличия зеараленона вторую ТСХ-пластинку опрыскивают 1 % раствором прочной синей В-соли или прочной фиолетовой В-соли в дистиллированной воде, затем пластинку немедленно опрыскивают 5 %-ным раствором углекислого натрия до появления красно-бордовой окраски пятен стандарта зеараленона. Обнаружение на пластинке пятна, соответствующего по цвету и хроматографической подвижности стандартам зеараленона, подтверждает наличие зеараленона в образце.
Количественное определение зеараленона с помощью двумерной ТСХ
Пластинку «Силуфол» размечают тонкими карандашными линиями, не повреждая слоя силикагеля, согласно рис. 30. В правом нижнем углу на расстоянии 1,5 см от краев пластинки наносят с помощью микрошприца 0,02 см3 раствора В. В правом верхнем углу наносят 0,003 и 0,007 см3 рабочего раствора зеараленона, 0,01 и 0,02 см3 раствора В; в левом нижнем углу наносят 0,005 и 0,01 см3 рабочего раствора зеараленона, 0,002 и 0,005 см3 раствора В. Пластинку помещают в камеру для ТСХ со смесью гексан - ацетон (7:3) и элюируют в первом направлении до достижения фронтом растворителя тонкой карандашной линии, проведенной в 5,5 см от верхнего края пластинки. Пластинку извлекают и сушат на воздухе 5 мин. Затем проводят хроматографию во втором направлении в системе толуол - этилацетат - хлороформ - 85 %-ная муравьиная кислота (45:25:25:5) до достижения карандашной линии, проведенной в 5,5 см от верхнего края пластинки. Пластинку извлекают из камеры, сушат и обнаруживают пятна по флуоресценции после обработки раствором хлорида алюминия, как описано выше. Сравнивая интенсивность флуоресценции разных количеств стандарта зеараленона с интенсивностью флуоресценции пятна зеараленона в экстракте (раствор В), определяют количество нг зеараленона в экстракте.
Содержание зеараленона в образце рассчитывают по формуле:
![]() ,
где
,
где
С - концентрация зеараленона в образце, мг/кг;
V1 - объем водно-ацетонитрильной смеси для экстракции, см3 (125 см3);
V2 - объем водно-ацетонитрильного фильтрата, взятый для анализа, см3 (50 см3);
V3 - объем очищенного экстракта перед ТСХ (раствор В), см3 (0,5 см3);
V4 - объем очищенного экстракта (раствор В), наносимый на пластинку, см3 (0,02 см3);
М - навеска образца, взятая для анализа, г (25 г);
m - масса зеараленона в V4 см3 очищенного экстракта, оцененная по ТСХ, нг;
K - степень извлечения зеараленона по табл. 54.
При приведенных в скобках объемах и навесках формула содержания зеараленона выражается следующим образом:
![]() ,
мг/кг.
,
мг/кг.
Если интенсивность флуоресценции пятна зеараленона в экстракте выше интенсивности флуоресценции пятна стандарта, соответствующего 0,02 см3 стандартного раствора (200 нг зеараленона), то на пластинку следует нанести либо меньшее количество экстракта (уменьшить объем V4) либо разбавить раствор В бензолом (увеличить объем V3), внеся соответствующие коррективы в расчетную формулу.
Обнаружение и количественное определение зеараленона с помощью ВЭЖХ
Условия и параметры анализа зеараленона с помощью ВЭЖХ приведены в табл. 53.
Таблица 53
Условия анализа зеараленона
|
Сорбент |
Подвижная фаза |
Расход подвижной фазы, см3/мин |
Примерный коэффициент емкости |
|
|
Нормальнофазный |
Силикагель |
Гексан-эфир-уксусная кислота (65:35:1) |
1,5 |
2,0 - 2,5 |
|
Нормальнофазный |
Силикагель |
Петролейный эфир-эфир-уксусная кислота (50:50:1) |
2 |
2,0 - 2,5 |
|
Обращеннофазный |
одс-силикагель С18 |
Метанол-вода-ацетонитрил (61:35:4) |
1 |
1,0 - 1,3 |
Рекомендуется использовать перегнанные гексан и метанол или петролейный эфир и эфир, профильтрованный через слой оксида алюминия (5 см). УФ-детектор устанавливают на длину волны 283 нм, шкала чувствительности 0,005 е.о.п. Шкала самописца 10 мВ. Если используют флуориметрический детектор, то длину волны на линии возбуждения устанавливают около 283 нм, а на линии эмиссии - фильтр от 420 нм (или 440 нм в случае монохроматора на линии эмиссии).
Для калибровки прибора в инжектор с помощью микрошприца вводят 0,002 и 0,005 см3 рабочего раствора зеараленона, что соответствует 20 и 50 нг зеараленона. Для каждого количества стандарта определяют высоту пика на хроматограмме.
В инжектор хроматографа вводят 0,01 см3 раствора В. При наличии пика, совпадающего по времени удерживания со стандартом, определяют его высоту (h).
Расчет концентрации зеараленона в образце проводят по формуле:
![]() ,
где
,
где
С - концентрация зеараленона в образце, мг/кг;
V1 - объем водно-ацетонитрильной смеси для экстракции, см3 (125 см3);
V2 - объем водно-ацетонитрильного фильтрата, взятый для анализа, см3 (50 см3);
V3 - объем очищенного экстракта перед ВЭЖХ (раствор В), см3 (0,5 см3);
V4 - объем очищенного экстракта (раствор В), внесенный в хроматограф, см3 (0,02 см3);
М - навеска образца, взятая для анализа, г (25 г);
т - масса стандарта зеараленона, введенная в хроматограф, нг;
hcm - высота пика, соответствующая данной массе стандарта, мм;
h - высота пика зеараленона из образца, мм;
K - степень извлечения зеараленона по табл. 54.
Если пик зеараленона в образце выходит за пределы шкалы самописца, анализ проводят повторно после разбавления раствора В бензолом, т.е. после увеличения объема V3.
При ВЭЖХ зеараленона чувствительность УФ- и флуориметрического детекторов примерно одинакова. Преимуществом флуориметрического детектора является его селективность (меньшее количество посторонних пиков на хроматограмме).
Метрологические характеристики
Относительное допустимое расхождение между результатами двух параллельных определений, выполненных в одной лаборатории, по отношению к среднему арифметическому значению (Rr) и относительное допустимое расхождение между результатами испытаний, выполненных в двух разных лабораториях, по отношению к среднему арифметическому значению (RR), а также предел определения по данным авторов методов анализа дезоксиниваленола и зеараленона приведены в табл. 54, в которой указаны также относительные внутрилабораторные (Rsr) и межлабораторные (RSR) среднеквадратичные отклонения и средняя степень извлечения, дезоксиниваленола (вомитоксина) и зеараленона.
Таблица 54
Относительные допустимые внутрилабораторные (Rr) и межлабораторные (RR) расхождения результатов определения, величина предела определения и степень извлечения для методов определения дезоксиниваленола и зеараленона
|
Уровень загрязнения, мг/кг |
Метод |
Rsr, % |
RSR, % |
Rr, % |
RR, % |
Предел определения, мг/кг |
Степень извлечения, % |
|
|
Дезоксиниваленол |
2,0 |
тСх |
29 |
46 |
81 |
129 |
0,150 |
80 |
|
ВЭЖХ |
11 |
20 |
31 |
56 |
0,060 |
80 |
||
|
0,4 |
тСх |
41 |
58 |
115 |
162 |
0,150 |
80 |
|
|
ВЭЖХ |
19 |
27 |
53 |
76 |
0,060 |
80 |
||
|
Зеараленон |
1,0 |
тСх |
26 |
48 |
73 |
134 |
0,060 |
85 |
|
вэжх-флуор. |
12 |
20 |
34 |
56 |
0,004 |
85 |
||
|
Зеараленон |
0,1 |
вэжх-УФ |
12 |
20 |
34 |
56 |
0,010 |
85 |
|
тСх |
44 |
54 |
132 |
152 |
0,060 |
85 |
||
|
вэжх-флуор. |
17 |
26 |
48 |
73 |
0,004 |
85 |
||
|
вэжх-УФ |
17 |
26 |
48 |
73 |
0,010 |
85 |
4. Метод обнаружения, идентификации и количественного определения патулина в БАД на плодоовощной основе
Подготовка проб
Навеску жидких БАД массой 50 г или фруктового порошка массой 25 г помещают в стеклянный стакан, смешивают с небольшим количеством дистиллированной воды и количественно переносят в мерную колбу вместимостью 250 см3. В мерную колбу вносят 15 см3 раствора Карреза I и 15 см3 раствора Карреза II. Содержимое колбы доводят дистиллированной водой до метки, тщательно перемешивают и фильтруют в мерный цилиндр через бумажный складчатый фильтр. Отбирают 50 см3 фильтрата.
Экстракция
Фильтрат из цилиндра переносят в делительную воронку, добавляют 50 см3 этилацетата и смесь интенсивно встряхивают. После разделения слоев отделяют верхний этилацетатный слой и переносят его в сухую плоскодонную колбу. Затем экстракцию проводят еще раз со свежей порцией этилацетата. Если полного расслоения жидкостей не происходит, в делительную воронку добавляют 10 - 12 г хлорида натрия и смесь слегка встряхивают. Этилацетатные экстракты объединяют, сушат безводным сульфатом натрия и фильтруют через кусочек ваты в грушевидную колбу. Сульфат натрия промывают 10 см3 этилацетата и фильтруют в ту же колбу. Экстракт упаривают на ротационном испарителе досуха. Остаток растворяют в 1 см3 этилацетата.
Очистка экстракта
В стеклянную хроматографическую колонку на дно помещают кусочек ваты и 5 см3 бензола, насыпают слой безводного сульфата натрия (5 мм) и наливают суспензию 2 г (3,6 см3) силикагеля в 15 см3 бензола. Не давая колонке просохнуть (колонка закрыта снизу), насыпают слой безводного сульфата натрия (10 мм). Бензолу дают стечь, на верхний слой сорбента наносят экстракт, дают впитаться в фильтрующий слой. Отгонную колбу ополаскивают 5 см3 этилацетата, переносят раствор на колонку и дают этилацетату полностью впитаться в фильтрующий слой. Колонку промывают 25 см3 бензола. Патулин элюируют 75 см3 смеси этилацетат - бензол (25:75). Элюат упаривают досуха на ротационном испарителе. Остаток растворяют в 0,2 см3 смеси бензол - ацетонитрил (9:1) для анализа методом ТСХ или в 0,2 см3 метанола для анализа методом ВЭЖХ (раствор А).
Обнаружение и количественное определение патулина с помощью двумерной ТСХ
Пластинку «Силуфол» размечают тонкими карандашными линиями согласно рис. 29. В правом нижнем углу на расстоянии 1,5 см от краев пластинки наносят с помощью микрошприца 0,02 см3 раствора А. В левом нижнем углу пластинки наносят 0,002 и 0,004 см3 стандартного раствора патулина, что соответствует 20 и 40 нг стандарта. В верхнем правом углу пластинки наносят 0,006 и 0,008 см3 стандартного раствора патулина, что соответствует 60 и 80 нг стандарта. Пластинку помещают в камеру для ТСХ со смесью хлороформ - ацетон (4:1) и элюируют в первом направлении до достижения фронтом растворителя верхней карандашной линии. Пластинку извлекают из камеры и сушат на воздухе. Затем проводят элюирование пластинки во втором направлении смесью толуол - этилацетат - муравьиная кислота (5:4:1). После достижения фронтом растворителя карандашной линии ее извлекают из камеры и сушат на воздухе до исчезновения запаха растворителя. На дно эксикатора помещают стеклянный стакан вместимостью 150 см3 с 25 см3 раствора марганцовокислого калия, осторожно приливают 25 см3 соляной кислоты, быстро устанавливают вставку эксикатора и на нее хроматографическую пластинку, плотно закрывают эксикатор. Через 15 мин пластинку извлекают из эксикатора и оставляют выветриваться в вытяжном шкафу еще на 15 - 20 мин до полного исчезновения запаха хлора. (Работу с хлором следует проводить в вытяжном шкафу лаборатории с использованием индивидуальных средств защиты). Затем пластинку опрыскивают раствором бензидина, выдерживают 20 - 30 мин и рассматривают в длинноволновом УФ-свете. Патулин обнаруживается в виде желтых флуоресцирующих пятен. Наличие на пластинке пятна, соответствующего по цвету флуоресценции и хроматографической подвижности пятнам стандарта патулина, свидетельствует о его наличии в анализируемом продукте. Сравнивая интенсивность флуоресценции пятна патулина в пробе (в растворе А) с интенсивностью флуоресценции стандартов, оценивают количество. Содержание патулина в анализируемом продукте определяют по формуле:
![]() ,
где
,
где
С - содержание патулина в продукте, мг/кг;
V1 - объем раствора, до которого доведена исходная навеска продукта, см (250 см3);
V2 - объем фильтрата, отобранный на анализ, см3 (50 см3);
V3 - объем очищенного хлороформного экстракта перед ТСХ, см3 (0,2 см3);
V4 - объем хлороформного экстракта, нанесенный на пластинку, см3 (0,02 см3);
т - количество патулина, обнаруженное в пятне, нг;
М - масса образца, взятая на анализ, г;
K - степень извлечения патулина по табл. 55.
Если интенсивность флуоресценции пятна патулина в экстракте выше интенсивности флуоресценции пятна стандарта, соответствующего 0,008 см3 стандартного раствора (80 нг патулина), то на пластинку следует либо нанести меньшее количество экстракта (уменьшить объем V4), либо разбавить раствор А хлороформом (увеличить объем V3), внеся соответствующие коррективы в расчетную формулу.
Обнаружение и количественное определение патулина с помощью ВЭЖХ
Условия ВЭЖХ: подвижная фаза изопропанол - гексан (1:4); расход подвижной фазы 1,0 см3/мин. Рекомендуется использовать перегнанные растворители. УФ-детектор устанавливают на длину волны 276 нм, шкала чувствительности 0,005 е.о.п. Шкала самописца 10 мВ.
Для калибровки прибора в инжектор с помощью микрошприца вводят 0,002; 0,005 и 0,015 см3 рабочего раствора патулина, что соответствует 4; 10 и 30 нг патулина. Для каждого количества стандарта определяют высоту пика на хроматограмме. При описанных условиях ВЭЖХ коэффициент емкости (К¢) для патулина составляет 2,3 - 2,7.
В инжектор хроматографа вводят 0,01 см3 раствора А. При наличии пика, совпадающего по времени удерживания со стандартом, определяют его высоту (hобр). Расчет концентрации патулина в образце проводят по формуле:
![]() ,
где
,
где
С - концентрация патулина в образце, мг/кг;
V1 - объем, до которого доведена навеска при экстракции водой, см3 (250 см3);
V2 - объем фильтрата, взятый для анализа, см3 (50 см3);
V3 - объем очищенного экстракта перед ВЭЖХ (раствор А), см3 (0,2 см3);
V4 - объем очищенного экстракта (раствор А), внесенный в хроматограф, см3 (0,02 см3);
М - навеска образца, взятая для анализа, г (50 или 25 г);
т - масса стандарта патулина, введенная в хроматограф, нг;
hcm. - высота пика, соответствующая данной массе стандарта, мм;
hобр. - высота пика патулина из образца, мм;
K - степень извлечения патулина по табл. 55.
Если пик патулина в образце выходит за пределы шкалы самописца, анализ проводят повторно после разбавления раствора А метанолом, т.е. после увеличения объема V3.
Метрологические характеристики
Относительное допустимое расхождение между результатами двух параллельных определений, выполненных в одной лаборатории, по отношению к среднему арифметическому значению (Rr) и относительное допустимое расхождение между результатами испытаний, выполненных в двух разных лабораториях, по отношению к среднему арифметическому значению (RR), а также предел определения по данным авторов методов анализа патулина приведены в табл. 55, в которой приведены также относительные внутрилабораторные (Rsr) и межлабораторные (RSR) среднеквадратичные отклонения и средняя степень извлечения патулина.
Таблица 55
Относительные допустимые внутрилабораторные (Rr) и межлабораторные (RR) расхождения результатов определения, величина предела определения и степень извлечения для методов определения патулина
|
Уровень загрязнения, мг/кг |
Метод |
Rsr, % |
RSR, % |
Rr, % |
RR, % |
Предел определения, мг/кг |
Степень извлечения, % |
|
0,1 |
тСх |
42 |
65 |
118 |
182 |
0,012 |
75 |
|
|
вэжх |
13 |
18 |
36 |
50 |
0,005 |
75 |
5. Метод обнаружения, идентификации и определения содержания трихотеценовых микотоксинов группы А в пищевых продуктах и БАД на зерновой основе с помощью газо-жидкостной хроматографии (арбитражный метод)
В основу положен метод групповой индикации трихотеценовых микотоксинов группы А, в т.ч. наиболее токсичного среди них Т-2 токсина, с помощью газожидкостной хроматографии по внутреннего стандарту - 3-О-метил-Т-2-тетраолу.
5.1. Экстракция
Навеску измельченной пробы зерна массой 20 г помещают в плоскодонную коническую колбу вместимостью 250 см3, добавляют 10 см3 4 %-ного раствора хлорида калия и 90 см3 ацетонитрила. Встряхивают на аппарате для встряхивания 30 мин. Полученную смесь фильтруют через бумажный складчатый фильтр в мерный цилиндр, отбирают 70 см3 фильтрата (соответствует 14 г исходного образца).
5.2. Очистка экстракта
В делительную воронку на 250 см3 помещают 70 см3 фильтрата, добавляют 50 см3 гексана, после встряхивания и разделения слоев верхний гексановый слой отбрасывают. Нижний ацетонитрильный слой еще дважды встряхивают с 40 см3 гексана, каждый раз отбрасывая верхний гексановый слой. Если слои плохо разделяются, добавляют сульфат аммония. К ацетонитрильному слою добавляют 2 - 3 см3 бензола и сульфат аммония на кончике шпателя, встряхивают и отбрасывают нижний водный слой. Ацетонитрильный слой помещают в круглодонную колбу и упаривают досуха с добавлением изопропанола до объема 1 см3.
5.3. Омыление пробы
К пробе добавляют 1 см3 4 %-ного раствора едкого натра в 30 %-ном изопропиловом спирте. Смесь выдерживают 2 ч при комнатной температуре.
5.4. Экстракция продуктов омыления
Щелочь нейтрализуют добавлением 2 %-ого раствора уксусной кислоты (около 2 см3) до рН среды 5 - 7 (контроль по универсальной индикаторной бумаге). Смесь помещают в делительную воронку или в пробирку вместимостью 10 см3, добавляют 3,5 см3 смеси ацетонитрил - бензол (3:0,5), на кончике шпателя сульфат аммония, встряхивают и отделяют в чистую сухую колбу верхний ацетонитрильный слой (при использовании пробирки экстракт отделяют с помощью пипетки). Экстракцию повторяют дважды. Объединенные экстракты высушивают добавлением 1 г безводного сульфата натрия, фильтруют через ватный тампон в грушевидную колбу, куда предварительно помещают около 200 мг силикагеля для колоночной хроматографии, и упаривают досуха на ротационном испарителе при температуре 40 - 45 °С (до получения сыпучего силикагеля).
5.5. Очистка продуктов омыления.
Очистку продуктов омыления проводят с помощью колоночной хроматографии. Для хроматографии используют растворители, очищенные путем дистилляции. Безводный сернокислый натрий и силикагель промывают последовательно бензолом и ацетоном, затем высушивают на воздухе и прокаливают в сушильном шкафу при 100 °С в течение часа.
На дно стеклянной колонки помещают кусочек ваты, 3 - 5 см3 бензола, безводный сульфат натрия (толщина слоя 5 мм, очищенный как указано выше), заливают суспензию 2 г (3,6 см3) силикагеля в бензоле, не давая бензолу стечь (колонка закрыта снизу), высыпают омыленную пробу на силикагеле по п. 5.4. и после оседания твердых частиц насыпают безводного сульфата натрия (10 мм). Дают бензолу стечь и колонку промывают последовательно 25 см3 смеси бензол - ацетон (95:5) (элюат отбрасывают) и 70 см3 смеси бензол-ацетон (1:1). Элюат собирают в круглодонную колбу, упаривают на ротационном испарителе до объема 1 см3. Остаток переносят в вайл (емкость вместимостью 4 - 6 см3 с герметично завинчивающейся крышкой) или в пробирку вместимостью 5 см3 с НШ 14,5, упаривают досуха в токе азота и остаток растворяют в 0,02 см3 бензола, очищенного дистилляцией над металлическим натрием (раствор А).
5.6. Получение ТФА-производных
5.6.1. Получение стандартного раствора 3-О-метил-Т-2-тетраола
5.6.1.1. Метелирование Т-2 токсина
1 г Т-2 токсина помещают в круглодонную колбу вместимостью 250 см3, добавляют 3 см3 очищенного перегонкой йодистого метила, 300 мг оксида серебра (катализатор) и кипятят на глицериновой бане (температура бани 60 °С) с обратным холодильником в течение 2 - 4 ч (контроль продуктов реакции по ТСХ на пластинках «Силуфол» в системе бензол - ацетон (5:1), эталон - исходный Т-2 токсин). После окончания реакции содержимое реакционной колбы охлаждают до комнатной температуры и упаривают досуха (от избытка йодистого метила) на ротационном испарителе. Остаток растворяют в 3 см3 бензола.
5.6.1.2. Очистка 3-О-Ме-Т-2-токсина от исходного Т-2 токсина
Продукт реакции 3-О-Ме-Т-2-токсин необходимо очистить от исходного Т-2-токсина с помощью колоночной хроматографии. Для этого на дно стеклянной колонки размером 300´22 мм помещают кусочек ваты, приливают 5 - 10 см3 бензола (колонка закрыта снизу), присыпают безводный сульфат натрия (толщина слоя 5 мм), заливают суспензию 20 г силикагеля в бензоле и насыпают слой безводного сульфата натрия (20 мм). Дают бензолу стечь и на колонку наносят раствор продуктов реакции в бензоле по п. 5.6.1.1. (3 см3). Реакционную колбу ополаскивают бензолом (0,5 см3) и раствор также наносят на колонку. Колонку последовательно промывают:
· бензолом - 100 см3,
· 2 %-ным раствором ацетона в бензоле - 200 см3,
· 4 %-ным раствором ацетона в бензоле - 100 см3,
· 5 %-ным раствором ацетона в бензоле - 600 см3,
· 10 %-ным раствором ацетона в бензоле - 300 см3,
· 15 %-ным раствором ацетона в бензоле - 200 см3.
Отбирают фракции, содержащие чистый 3-О-Ме-Т-2-токсин (5 %-ная смесь ацетона в бензоле) и Т-2 токсин (10 - 15 %-ная смесь ацетона в бензоле). Контроль реакции проводят с помощью ТСХ на пластинках «Силуфол» в системе бензол - ацетон (5:1). Выход 3-О-Ме-Т-2-токсина после упаривания растворителя досуха на ротационном испарителе и высушивания остатка в вакууме составляет порядка 0,5 г (50 %). Возврат Т-2-токсина - 50 %.
5.6.1.3. Омыление Ме-Т-2-токсина.
К высушенному остатку 3-О-Ме-Т-2-токсину добавляют 10 см3 метанола, 2 см3 воды и 3 см3 раствора 3Н К2СО3. Смесь нагревают 2 ч при 40 °С при перемешивании на магнитной мешалке. Контроль продуктов реакции проводят с помощью ТСХ на пластинках «Силуфол» в системе бензол - ацетон (1:1). По окончании реакции смесь охлаждают, разбавляют 70 см3 воды и нейтрализуют добавлением 0,4 см3 ледяной уксусной кислоты до слабо кислой среды (контроль по универсальной индикаторной бумажке).
5.6.1.4. Очистка 3-О-Ме-Т-2-тетраола на патроне Supelclean LC-18.
Полученный раствор по п. 5.6.1.3. пропускают через концентрирующий патрон Supelclean LC-18 (толщина слоя 1 см), элюат отбрасывают. Патрон промывают 50 см3 метанола. Элюат содержит неполярные примеси и следовые количества продукта и его отбрасывают. 3-О-Ме-Т-2-тетраол смывают 10 см3 смеси метанол - вода (5:1) и экстрагируют этилацетатом (5 см3 ´ 3 раза). При плохом расслоении жидкостей в элюат добавляют хлорид натрия или центрифугируют. Этилацетатные экстракты объединяют, упаривают досуха и 3-О-Ме-Т-2-тетраол кристаллизуют из бензола с добавлением петролейного эфира до помутнения. Выход 3-О-Ме-Т-2-тетраол составляет порядка 70 %.
5.6.1.5. Приготовление стандартного рабочего раствора 3-О-Метил-Т-2-тетраола.
625 мг 3-О-Ме-Т-2-тетраола помещают в мерную колбу вместимостью 25 см3, содержащую до половины бензол, перемешивают до полного растворения вещества и доводят бензолом до метки, тщательно перемешивают. Концентрация 3-О-Ме-Т-2-тетраола в полученном растворе 25 мг/см3.
5.6.2. Получение трифторацетилъных производных стандартной смеси Т-2-тетраола и 3-О-Метш-Т-2-тетраола
В вайл (емкость вместимостью 4 - 6 см3 с герметично завинчивающейся крышкой) или пробирку вместимостью 5 см3 с НШ 14,5 помещают 0,02 см3 рабочего раствора Т-2 тетраола (1 мкг) и 0,04 см3 рабочего раствора 3-О-метил-Т-2-тетраола (1 мкг) в бензоле, добавляют 0,1 см3 бензола, предварительно очищенного дистилляцией над металлическим натрием, 30 - 50 мг свежепрокаленного углекислого натрия и 0,05 см3 трифторуксусного ангидрида. Пробирку плотно закрывают стеклянной притертой пробкой и нагревают при 60 °С в течение 0,5 часа при периодическом встряхивании. Содержимое пробирки разбавляют бензолом до 1 см3 и фильтруют через химическую воронку с кусочком ваты в другую пробирку вместимостью 5 см3 с НШ 14,5. Углекислый натрий на фильтре промывают 0,5 см3 бензола. Объединенные бензольные фильтраты упаривают досуха в токе азота (при отсутствии баллона с азотом можно упарить досуха на ротационном испарителе). Остаток растворяют в 0,02 см3 бензола и анализируют с помощью ГЖХ.
5.6.3. Получение ТФА-производных продуктов омыления.
К 0,02см3 раствора продуктов омыления экстракта по п. 5.5. (раствор А) добавляют 0,04 см3 стандартного раствора 3-О-метил-Т-2-тетраола (1 мкг, внутренний стандарт) в бензоле, 30 - 50 мг свежепрокаленного углекислого натрия и 0,05 см3 трифторуксусного ангидрида. Пробирку плотно закрывают стеклянной притертой пробкой и проводят получение ТФА-производных согласно п. 5.6.2 при периодическом встряхивании.
5.7. ГЖХ анализ ТФА производных
Условия ГЖХ анализа: колонка капиллярная, длина колонки 25 м, жидкая фаза SE-54, температура колонки - 210 °С, температура термостата детектора - 240 °С, температура инжектора - 300 °С, скорость газа-носителя 2 см3/мин, скорость продувочного газа - 6 см3/мин, шкала чувствительности - 2´10-12 А (на блоке ИМТ).
В инжектор газового хроматографа с помощью микрошприца вместимостью 1 или 10 мкл последовательно вводят две аликвоты (около 0,2 - 0,5 мкл) стандартной смеси ТФА-производных Т-2-тетраола и 3-О-метил-Т-2-тетраола по п. 5.6.2. При использовании метода внутреннего стандарта нет необходимости вводить в хроматограф точные количества проб. В каждом случае регистрируют время выхода растворителя бензола и время выхода ТФА-Т-2 тетраола и ТФА-3-О-метил-Т-2-тетраола. Определяют относительное приведенное время удерживания токсина (наиболее воспроизводимая величина для идентификации) и площади пиков токсинов (Sст. и Sвн.ст.). Относительные приведенные времена удерживания определяют по следующей формуле:
![]() ,
где
,
где
![]() - относительное приведенное время удерживания, мин.,
- относительное приведенное время удерживания, мин.,
t0 - время удерживания растворителя, мин.,
![]() - время удерживания Т-2 тетраола, мин.,
- время удерживания Т-2 тетраола, мин.,
![]() - время удерживания 3-О-метил-Т-2-тетраола, мин.
- время удерживания 3-О-метил-Т-2-тетраола, мин.
Площадь пиков определяют по показанию интегратора или умножением высоты пика на ширину пика на половине его высоты. Затем в газовый хроматограф вводят 0,2 - 0,5 мкл раствора ТФА производного продуктов омыления экстракта по п. 5.6.3. При наличии пика, соответствующего относительному приведенному времени удерживания ТФА-производному Т-2 тетраола, определяют его площадь (Sобр.).
Относительное приведенное время удерживания Т-2 тетраола в зависимости от условий хроматографии может меняться в пределах 0,59 - 0,62.
5.8. Обработка результатов анализа
Расчет содержания Т-2 тетраола в анализируемой смеси проводят по формуле:
![]() ,
где
,
где
М - масса анализируемого вещества (Т-2 тетраола), мкг,
![]() - масса введенного внутреннего стандарта
(3-О-метил-Т-2-тетраола), мкг,
- масса введенного внутреннего стандарта
(3-О-метил-Т-2-тетраола), мкг,
S - площадь пика анализируемого вещества (Т-2 тетраола), мм2,
![]() - площадь пика внутреннего стандарта (3-О-метил-Т-2-тетраола),
мм2,
- площадь пика внутреннего стандарта (3-О-метил-Т-2-тетраола),
мм2,
K - поправочный коэффициент вещества (Т-2 тетраола) по внутреннему стандарту (3-О-метил-Т-2-тетраолу).
Поправочный коэффициент определяют в соответствии с параметрами калибровочной смеси по следующей формуле:
![]() ,
где
,
где
m - масса Т-2 тетраола, нг,
![]() - масса 3-О-метил-Т-2 тетраола, нг,
- масса 3-О-метил-Т-2 тетраола, нг,
![]() - площадь 3-О-метил-Т-2 тетраола, мм2,
- площадь 3-О-метил-Т-2 тетраола, мм2,
S - площадь Т-2 тетраола, мм2.
Пересчет количества Т-2 тетраола на кг исследуемого образца проводят следующим образом:
![]() , где
, где
![]() - содержание суммы трихотеценовых микотоксинов группы
А (Т-2 токсин, НТ-2 токсин, Т-2 тетраол и др.), мкг/кг,
- содержание суммы трихотеценовых микотоксинов группы
А (Т-2 токсин, НТ-2 токсин, Т-2 тетраол и др.), мкг/кг,
М - масса анализируемого вещества (Т-2 тетраола), определенная во вколе, мкг,
m - масса анализируемой пробы, соответствующая 70 см3 фильтрата (14 г) по п. 5.1., г.
5.9. Метрологические характеристики метода.
Предел обнаружения Т-2 тетраола во вколе при ГЖХ с использованием детектора электронного захвата (ДЭЗ) - 0,5 - 1 нг; общий предел обнаружения 20 мкг/кг (0,02 мг/кг); относительное стандартное отклонение 0,28 - 0,35 (при уровне загрязнения 0,1 мг/кг).
II. Метод определения нитратов и нитритов
Методы определения нитратов и нитритов распространяется на БАД на растительной основе, определение проводится либо методом фотометрии в соответствии с ГОСТ 8558.1-78, либо методом ионометрии по ГОСТ 29270-95 «Продукты переработки плодов и овощей. Методы определения нитратов и нитритов».
Рис. 28. Разметка пластины для одномерной ТСХ.
ст. 1, ст. 2, ст. 3 - точки нанесения рабочих растворов микотоксинов; А - точки нанесения раствора А.
Рис. 29. Разметка пластины «Силуфол» для двумерной ТСХ микотоксинов.
ст. 1, ст. 2, ст. 3, ст. 4, ст. 5 - точки нанесения рабочих растворов микотоксинов; А - точка нанесения раствора А.
Рис. 30. Разметка пластины «Силуфол» для двумерной ТСХ дезоксиниваленола и зеараленона.
ст. 1, ст. 2, ст. 3 - точки нанесения стандартных растворов микотоксинов; А - точки нанесения раствора А.
III. Метод определения n-нитрозаминов
Для определения N-нитрозаминов используют ГЖХ-хемилюминесцентный метод. Метод идентификации и количественного определения НА состоит в выделении летучих НА путем перегонки с паром или в вакууме, экстракции хлористым метиленом НА из водного дистиллята, концентрировании экстракта, разделении смеси методом газожидкостной хроматографии и количественном определении немодифицированных НА с помощью высокоселективного и высокочувствительного хемилюминесцентного (термоэнергетического) детектора ТЕА-502.
Идентификацию НА осуществляют по времени удерживания в сравнении с параметрами удерживания стандартных НА. Количественное определение проводят методом абсолютной калибровки с систематическим контролем калибровочного коэффициента.
1. Аппаратура, материалы, реактивы
Весы технические и весы аналитические.
Шкаф сушильный, нагревательные приборы (электронагреватель, колбонагреватель или электроплитка).
Ротационный испаритель с ловушкой.
Насос водоструйный.
Контактные термометры.
Микрошприц МШ-10 на 10 мкл или калиброванные стеклянные капилляры.
Система для отгонки нитрозаминов с водяным паром, принципиальная схема которой представлена на рис. 23.
Газовый хроматограф любой марки.
Детектор хемилюминесцентный - анализатор термической энергии типа ТЕА-502 фирмы «Termo Electron Corporation» (США).
Самописец - любой марки.
Колонка газохроматографическая стеклянная длиной 3 м и диаметром 2 мм, заполненная 15 % Carbowax-20M, нанесенном на Chromaton N-AW-DMCS (80 - 100 меш.).
Азот газообразный (ос.ч. или поверочный нулевой газ) или аргон газообразный, ос.ч.
Кислород газообразный, ос.ч.
Азот жидкий.
N-нитрозодиметиламин (НДМА).
N-нитрозодиэтиламин (НДЭА).
N-нитрозодипропиламин (НДПА).
N-нитрозодибутиламин (НДБА).
N-нитрозопиперидин (НПип).
N-нитрозопирролидин (НПир) фирмы «Fluka» (Швейцария).
Гексановый раствор НДПА (0,2 мкг/см3) - внутренний стандарт.
Гексановый раствор смеси стандартных НА (НДМА, НДЭА, НДБА, НПиП, НПир) по 0,2 мкг каждого НА в 1 см3 раствора.
Ацетонитрил, х.ч.
Бензол, х.ч.
Вода дистиллированная.
н-Гексан, х.ч.
Кальция хлорид прокаленный, ч.
Кислота соляная, х.ч., и ее водный раствор 0,1 н.
Кислота серная, х.ч., и ее водные растворы 1 н. и 5 н.
Кислота уксусная ледяная, х.ч.
Метилена хлорид, х.ч.
Натрия гидроокись, х.ч., и его водный раствор 0,1 н.
Натрий сернокислый прокаленный, х.ч.
Натрий углекислый кислый, х.ч.
Натрий хлористый, х.ч.
Спирт этиловый.
2. Требования техники безопасности при проведении испытаний
Помещение, в котором производят определение нитрозаминов, обязательно должно быть оборудовано приточно-вытяжной вентиляцией.
Работу с нитрозаминами, стандартами, другими химическими соединениями и растворителями проводить в вытяжном шкафу с использованием индивидуальных средств защиты (очки, перчатки и др.).
В лаборатории, где проводится работа с канцерогенными летучими НА, необходимо всегда иметь 2 - 3 %-ный раствор газообразного НВr в уксусной кислоте для разрушения НА при попадании их на рабочие места и пол. В целях разрушения летучих НА в воздухе по окончании работы помещение необходимо обработать ультрафиолетовым светом.
Транспортировка и хранение НА осуществляются в стеклянных запаянных ампулах, обернутых в асбестовую ткань и упакованных в металлическую тару, которую также заплавляют парафином.
НА хранят в специальном холодильнике в отсутствие анализируемых проб.
1,0 см3 раствора НДПА из ампулы с концентрацией 0,2 мг/см3 переносят в мерную колбу на 100 см3 и доводят гексаном до метки. Полученный раствор с концентрацией 2 мкг/см3 используют при анализе в качестве внутреннего стандарта.
3. Выделение нитрозаминов
3.1. Выделение нитрозаминов путем перегонки с паром
Навеску 50 - 100 г измельченного в гомогенизаторе БАД помещают в круглодонную колбу на 500 см3, соединенную с паровиком и прямым холодильником (принципиальная схема установки представлена рис. 31). К образцу добавляют 10 г хлористого натрия, 10 г сульфата натрия или магния, 25 - 50 см3 дистиллированной воды (в зависимости от влажности образца), 5 см3 2 % раствора сульфаминовой (или сульфаниловой) кислоты, 5 - 10 см3 1 н. раствора серной кислоты до рН < 3,0; 1 см3 гексанового раствора ДПНА и НА отгоняют с водяным паром, собирая 200 - 230 см3 дистиллята. Если перегонка с паром сопровождается вспениванием массы, добавляют 5 - 10 см3 насыщенного раствора апиезона в изопропиловом спирте, а в случае спекания образцов в процессе дистилляции - мелкие керамические крошки или кусочки стеклянных трубочек. Для наиболее полного выделения НА из БАД, содержащих спирт, необходимо разбавить образец водой до получения 20 % концентрации в нем спирта. Дистиллят переносят в делительную воронку и нитрозамины экстрагируют свежеперегнанным хлористым метиленом 4 - 5 раз по 15 см3.
Объединенные хлорметиленовые экстракты высушивают прокаленным сульфатом магния, отфильтровывают, осушитель промывают небольшим объемом хлористого метилена, объединенные фильтраты помещают в круглодонную колбу на 100 - 150 см3, снабжают дефлегматором (или воздушным холодильником), помещают в водяную баню и упаривают хлористый метилен до 5 - 7 см3 выдерживанием при температуре бани 55 - 65 °С. Целесообразно пропускание тока инертного газа через хлорметиленовый раствор. По достижении объема раствора 5 - 7 см3 дефлегматор смывают 2 см3 гексана и продолжают концентрировать пробу до 2 см3.
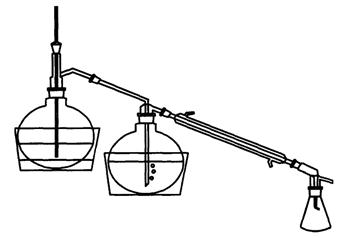
Рис. 31. Система для отгонки нитрозаминов с водяным паром.
3.2. Выделение нитрозаминов перегонкой в вакууме
Пробу исследуемого образца массой 20 - 50 г, отвешенную с погрешностью 0,1 г, помещают в круглодонную колбу вместимостью 200 см3, добавляют 75 - 80 см3 дистиллированной воды, 1 см3 гексанового раствора НДПА, 4 см3 0,1 н. раствора гидроокиси натрия и 20 см3 парафинового или вазелинового масла для предотвращения перебрасывания массы во время дистилляции и присоединяют к вакуумной системе (принципиальная схема установки изображена на рис. 32).
Колбу с исследуемым образцом помещают на песочную баню, под которой устанавливают газовую горелку; приемную колбу охлаждают смесью сухого льда и гексана (ацетона) или жидким азотом. Установив в системе вакуум до 0,1 атм, проводят дистилляцию до получения 50 см3 дистиллята. Чтобы избежать потери НА, обязательно проводят дистилляцию в закрытой системе. Герметичность системы можно сохранить, используя чистую вакуумную смазку или тефлоновые манжеты для шлифов. После окончания дистилляции снимают обогрев, охлаждение, отсоединяют шланг вакуумной системы. После оттаивания приемной колбы систему перегонки разбирают, насадку ополаскивают несколькими см3 дистиллированной воды, которые добавляют к дистилляту. В круглодонную колбу с дистиллятом добавляют 1 - 2 см3 этанола, 0,5 см3 5 н. серной кислоты и экстрагируют трижды по 7 - 10 см3 хлористым метиленом. Экстракт высушивают прокаленным сульфатом магния и концентрируют аналогично п. 3.1 до 2 см3.
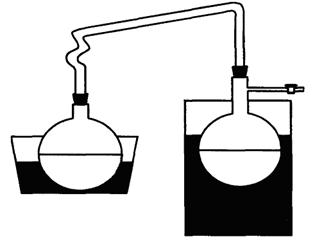
Рис. 32. Система вакуумной установки для перегонки нитрозаминов.
4. Подготовка к испытанию и проведение анализа
Анализ НА осуществляют с помощью ГЖХ без дополнительной обработки хлорметиленового или гексанового раствора при следующих условиях: колонка стеклянная (длина 3 м с диаметром 2 мм), заполненная 15 % Carbowax-20M, нанесенном на Chromaton N-AW-DMCS (80 - 100 меш.). (Может быть использована другая по размеру набивная колонка, например, с содержанием Carbowax 10 %), газ-носитель - азот или аргон, скорость 20 см3/мин, температура колонки 125 °С (изотермический режим); температура испарителя 220 °С, температура каталитической печи 450 °С, давление азота - 0,5 атм (или расход - 40 см3/мин), давление кислорода - 0,6 атм, температура охлаждающей ловушки 110 - 130 °С, объем вводимой в хроматограф пробы 1 - 10 мкл.
Записывают хроматограмму эталонной смеси НА и раствора внутреннего стандарта в начале и в конце каждой серии анализов. Идентификацию НА в пробе осуществляют по времени удерживания стандартов. Количество НА в пробе оценивают сравнением величины аналитических сигналов, полученных при анализе образцов и стандартных растворов.
Оценивают также полноту извлечения НА из БАД по внутреннему стандарту (НДПА), вносимому в продукт перед выделением НА.
Содержание НА в пробе рассчитывают по формуле:
![]() ,
где
,
где
С - содержание исследуемого НА в образце, мкг/кг;
S1 - площадь под интегральной кривой, полученной при анализе образца, мм2;
S2 - площадь под интегральной кривой соответствующего стандартного НА, мм2;
V1 - объем пробы, введенной в хроматограф, мкл;
V2 - объем анализируемого экстракта, мкл;
п - количество стандартного НА, введенного в хроматограф, нг;
т - масса пробы, взятой на анализ, г;
А - степень извлечения внутреннего стандарта (70 - 95 %).
5. Метрологические характеристики
Относительное допустимое расхождение между результатами двух параллельных определений, выполненных в одной лаборатории по отношению к среднему арифметическому значению (Rr) и относительное допустимое расхождение между результатами испытаний, выполненных в двух разных лабораториях, по отношению к среднему арифметическому значению (RR), а также предел определения приведены в табл. 56.
Таблица 56
Относительные допустимые внутрилабораторные (Rr) и межлабораторные (RR) расхождения результатов определения и величина предела определения
|
Предел определения, мкг/кг |
Rr, % |
RR, % |
|
|
Хемилюминесцентный |
0,1 |
20 |
45 |
IV. Метод определения биогенных аминов
1. Колориметрический метод определения гистамина
Принцип метода
В основе колориметрического метода определения гистамина лежит измерение величины абсорбции окрашенного производного, при взаимодействии гистамина с диазореактивом. Предел обнаружения - 10 мг/кг, относительное стандартное отклонение при определении гистамина в интервале 20 - 75 мг/кг изменяется от 0,08 до 0,25, степень извлечения добавленного к образцу стандарта гистамина - 94 - 99 %.
Специфические приборы, реактивы, материалы
Колориметр фотоэлектрический по ГОСТ 12083-78 со светофильтром = 490 + 10 нм, спектрофотометр СФ - 26 или аналогичный.
Микроизмельчитель тканей РТ - 2 по МРТУ 64.1-1505-63.
Гистамин или гидрорхлорид гистамина.
Кислота соляная по ГОСТ 3118-77, х.ч. раствор 3,75 г/л (0,1 н раствор).
Кислота трихлоруксусная по ТУ 6-09-1926-77, раствор 50 г/л (5 % раствор).
Натрий азотистокислый по ГОСТ 4197-77, раствор 50 г/л (5 % раствор).
Натрий гидроокись по ГОСТ 4328-77,ч.д.а., раствор 200г/л (5 % раствор).
Натрий сернокислый безводный по ГОСТ 4166-76, х.ч., прокаленный.
Натрий углекислый по ГОСТ 83-79, х.ч., раствор 40 г/л (4 % раствор).
Пара-нитроанилин, ч.д.а.
Этилацетат по ГОСТ 22300-76, ч.д.а.
Подготовка к испытанию
Приготовление раствора пара-нитроанилина
Пара-нитроанилин очищают путем перекристаллизации в воде. Для этого его растворяют в горячей дистиллированной воде и охлаждают раствор до комнатной температуры. Выпавшие кристаллы отфильтровывают и высушивают при 80 °С в течение 10 - 15 мин. 0,1 г пара-нитроанилина растворяют в 100 см3 0,1 н соляной кислоты. Хранят раствор в холодильнике до момента употребления.
Приготовление диазореактива
Диазореактив получают непостредственно перед использованием путем смешивания 10 см3 раствора 1 г/дм3 пара-нитроанилина в 0,1 н соляной кислоте и 1 см3 раствора 50 г/дм3 азотистокислого натрия и охлаждают до 0 °С.
Приготовление н-бутанола, насыщенного водой
Для приготовления н-бутанола, насыщенного водой, встряхивают 50 см3 н-бутанола и 20 см3 дистиллированной воды в делительной воронке. После разделения фаз отделяют верхний бутанольный слой.
Экстракция
Навеску 10 г (с точностью до 0,01 г) приготовленного образца помещают в сосуд микроизмельчителя тканей, добавляют 25 мл 5 % раствора трихлоруксусной кислоты и перемешивают 5 мин. Полученную смесь переносят в плоскодонную коническую колбу на 100 см3, ополаскивают сосуд смесителя дважды 5 - 10 см3 5 %-ного раствора трихлоруксусной кислоты и растворы объединяют. Колбу снабжают воздушным холодильником и выдерживают на водяной бане при 60 °С в течение 15 мин. Охлажденную смесь переносят количественно в цилиндр, доводят до 50 см3 5 %-ным раствором трихлоруксусной кислоты и фильтруют через складчатый фильтр (фильтрат 1).
Построение калибровочной кривой
Готовят стандартные растворы гистамина с концентрацией 5, 10, 20, и 40 мкг/см3 в 5 %-ной трихлоруксусной кислоте. Для приготовления основного раствора гистамина с концентрацией 40 мкг/см3 - 4,0 мг гистамина помещают в мерную колбу вместимостью 100 см3, растворяют в 5 %-ной трихлоруксусной кислоте и доводят объем до метки. Рабочие растворы гистамина с концентрациями 20, 10 и 5 мкг/см3 получают путем разбавления 5 %-ным раствором трихлоруксусной кислоты основного раствора в 2, 4 и 8 раз соответственно. Основной раствор гистамина хранят в холодильнике неделю, рабочие растворы готовят в день проведения анализа.
5 см3 каждого стандартного раствора помещают в пробирки с притертыми пробками, добавляют 1 см3 20 %-ного раствора гидроксида натрия и вносят в раствор при перемешивании безводный углекислый натрий до получения насыщенного раствора. Добавляют 5 см3 н-бутанола, насыщенного водой. Энергично встряхивают пробирки в течение 30 сек. После разделения фаз отбирают пипеткой или шприцем 3 см3 верхнего бутанольного слоя и переносят в другую пробирку с притертой пробкой, содержащую 3 см3 0,1 н раствора соляной кислоты. Встряхивают содержимое пробирки в течение 30 сек. После разделения фаз отбирают 2 см3 нижнего водного слоя в другую пробирку. Добавляют 2 см3 4 %-ного раствора углекислого натрия и выдерживают 5 мин при 0 °С. Добавляют 4 см3 этилацетата и энергично встряхивают в течение 30 сек. После разделения фаз сухой пипеткой отбирают верхний слой и переносят в пробирку, содержащую безводный сульфат натрия (раствор окрашенного производного в этилацетате должен быть прозрачным). Измеряют величину абсорбции раствора при 495 нм в кювете толщиной 1 см. В качестве раствора сравнения используют этилацетат.
На основании полученных данных строят калибровочную кривую зависимости абсорбции от концентрации гистамина в растворе.
Проведение испытаний
5 см3 фильтрата 1 помещают в пробирку и проводят процедуру, описанную выше, начиная с добавления раствора гидроокиси натрия.
Для определения концентрации гистамина в экстракте используют калибровочную кривую.
Обработка результатов
Содержание гистамина в образце (мг/кг) вычисляют по формуле:
![]() ,
где
,
где
с - концентрация гистамина, найденная по калибровочной кривой, мкг/см3;
V - объем экстракта, см3 (50 см3);
т - навеска образца БАД, г (10 г);
Ф - фактор разбавления:
![]() .
.
Вычисления проводят до единиц. За окончательный результат принимают среднее арифметическое значение результатов трех параллельных определений.
Метрологические характеристики
Допустимое расхождение между результатами параллельных определений не должно превышать 20 % по отношению к среденеарифметическому значению.
2. Определение содержания биогенных аминов с помощью ВЭЖХ
Принцип метода
Метод определения содержания биогенных аминов устанавливает хроматографическую методику выполнения измерений массовых концентраций биогенных аминов (тирамина, кадаверина, путресцина, спермидина, спермина, гистамина) в биологически активных добавках к пище и предназначены для проведения лабораторных исследований пищевой продукции и биологически активных добавок в соответствии с СанПиН 2.3.2.1078-01 «Гигиенические требования безопасности и пищевой ценности пищевых продуктов».
Специфические приборы, реактивы, материал
Жидкостной хроматограф с насосом высокого давления с подачей растворителя от 0,1 до 5,0 см3/мин., оборудованный спектрофотометрическим детектором с переменной длиной волны и системой для сбора и обработки хроматографических данных Мультихром, версия 1,5х (Амперсенд, Россия).
Колонка и предколонка хроматографические с силикагелем с размером частиц 5 мкм; длина колонки - 25 см, предколонки - 4,5 см, внутренний диаметр колонок - 0,46 см;
Микрошприцы МШ-10 и МШ-25 для жидкостной хроматографии.
Микроизмельчитель тканей РТ - 2 по МРТУ 64.1-1505-63.
Аппарат для встряхивания проб типа АВУ-6С, ТУ 64-1-2451-78.
Весы лабораторные общего назначения по ГОСТ 24104 с наибольшим пределом взвешивания 200 г и погрешностью ±0,0001 г.
Весы лабораторные с наибольшим пределом взвешивания до 200 г, с пределом допустимой погрешности ±0,0005 г по ГОСТ 2404-80.
Колбы мерные наливные 2-50-2, 2-100-2 по ГОСТ 1770.
Центрифуга лабораторная.
Пипетки 4-1-2 или 5-1-2, 4-2-10 по ГОСТ 29227.
Кислота трихлоруксусная по ТУ 6-09-1926-77, раствор 50 г/л (5 %-ный раствор).
Кислота соляная по ГОСТ 3118, х.ч., раствор 0,1Н.
Натрий сернокислый безводный по ГОСТ 4166-76, х.ч., прокаленный.
Натрий углекислый по ГОСТ 83-79, х.ч., насыщенный раствор.
Натрий хлористый по ГОСТ 4233-77, х.ч., насыщенный раствор.
Этилацетат по ГОСТ 22300-76, ч.д.а.
Гексан по ТУ 6-09-3375-73, ч.
Бензол для хроматографии, х.ч. по ТУ 6-09-779-76.
Ацетон по ГОСТ 2603-71, ч.д.а.
5-диметиламино-нафталинсульфонил хлорид (дансилхлорид), Serva, раствор 5 г/л в ацетоне.
Фильтры обеззольные ФО-ФС-15 «Синяя лента» по ТУ 2642-001-42624157-98.
Фильтр нейлоновый имп. на шприце в обойме 25 мм, размер пор 0,45 мкм.
Вода дистиллированная, ГОСТ 6709.
Баллон с сжатым азотом, ос.ч. по ГОСТ 9293-74.
Тирамин, кадаверин, путресцин, спермидин, спермин, гистамин или их гидрохлориды.
Подготовка к испытанию
Приготовление основных стандартных растворов биогенных аминов (БА)
Навески по 0,25 г аминов или соответствующие навески аминов гидрохлоридов помещают в мерные колбы вместимостью 50 см3, растворяют в 0,1М растворе соляной кислоты, доводят этим же раствором до метки и тщательно перемешивают. Получают стандартный раствор аминов в 0,1М HCl с массовыми долями каждого амина (тирамин, кадаверин, путресцин, спермидин, спермин, гистамин) 5 мг/см3;
Приготовление рабочих стандартных растворов биогенных аминов
Аликвоты в 1 см3 основного стандартного раствора каждого биогенного амина переносят в мерные колбы вместимостью 50 см3, доводят 0,1М HCl до метки и тщательно перемешивают. Получают рабочие стандартные растворы биогенных аминов концентрацией 0,1 мкг/см3.
Получение стандартных растворов ДНС-производных биогенных аминов
Для получения стандартных растворов ДНС-производных биогенных аминов отбирают по 0,05 см3 каждого рабочего стандартного раствора БА (5 мкг амина), помещают в вайл вместимостью 4 - 6 см3, добавляют 0,5 см3 насыщенного раствора бикарбоната натрия (рН 8,5 - 9) и 1 см3 ацетонового раствора ДНС-хлорида с концентрацией 5 г/л. Смесь перемешивают и выдерживают при комнатной температуре 12 - 14 ч или 1 ч при 45 - 50 °С.
В реакционную смесь добавляют 1 см3 дистиллированной воды и отдувают ацетон в токе азота. При этом можно вайл поместить в водяную баню, нагретую до 35 - 40 °С. ДНС-амиды экстрагируют бензолом (3´1 см3), каждый раз стеклянной пипеткой отбирая верхний бензольный слой. Объединенные бензольные экстракты сушат безводным сульфатом натрия, декантируют в сухой вайл, осушитель промывают бензолом, также декантируют и упаривают досуха. Перед анализом стандартные растворы ДНС-производных биогенных аминов растворяют в 1 см3 бензола.
Подготовка проб к анализу
Экстракция
Навеску 20 г пищевого продукта, взятую с точностью до 0,01 г, размельченного в мясорубке или гомогенизаторе, или 20 - 40 таблеток или капсул БАД (в зависимости от веса таблетки), размельченных в ступке, помещают в коническую колбу вместимостью 250 см3. Добавляют, соответственно, 100 см3 и 50 см3 5 %-ного раствора трихлоруксусной кислоты, снабжают воздушным холодильником, перемешивают и выдерживают 15 мин при температуре 60 °С на водяной бане, периодически встряхивая содержимое колбы. Полученную смесь охлаждают и фильтруют через складчатый фильтр.
Получение дансильных производных биогенных аминов из образца
Аликвоту экстракта 0,5 см3, соответствующую 0,1 г исходного образца, помещают в вайл вместимостью 4 - 6 см3, добавляют 0,5 см3 насыщенного раствора бикарбоната натрия (рН 8,5 - 9) и 1 см3 ацетонового раствора ДНС-хлорида с концентрацией 5 г/л. Смесь перемешивают и выдерживают при комнатной температуре 12 - 14 ч или 1 ч при 45 - 50 °С.
В реакционную смесь добавляют 1 см3 насыщенного раствора натрия хлорида и отдувают ацетон в токе азота. При этом можно вайл поместить в водяную баню, нагретую до 35 - 40 °С. ДНС-амиды экстрагируют бензолом (3´1 см3), каждый раз стеклянной пипеткой отбирая верхний бензольный слой. Если расслоение жидкостей происходит трудно, смесь центрифугируют в лабораторной центрифуге. Объединенные бензольные экстракты сушат безводным сульфатом натрия, декантируют в сухой вайл, осушитель промывают бензолом, также декантируют и упаривают досуха. Перед анализом сухой остаток растворяют в 1 см3 бензола.
Проведение испытаний
Условия хроматографического анализа
Насос высокого давления с верхним пределом давления не менее 25 МПа, диапазоном регулирования подачи растворителя не менее 0,1 - 5,0 см3/мин.
Петлевое устройство ввода проб с рабочим объемом петли 0,020 см3.
Флуориметрический детектор, длина волны возбуждающего излучения 335 нм, длина волны эмиссии 470 нм, проточная кварцевая кювета объемом 0,015 - 0,025 см3, с уровнем флуктуационных шумов не более 8´10-5 ед. флуоресценции, относительной погрешностью измерения интенсивности флуоресценции не более 2 %.
Подвижная фаза гексан - этилацетат 55:45.
Скорость потока 1,0 мл/мин.
Выполнение измерений
В инжектор хроматографа микрошприцем вводят 0,005 см3 каждого стандартного раствора ДНС-производного биогенных аминов и определяют времена удерживания и площади пиков на хроматограмме. Градуировку проводят через каждые 6 часов работы прибора.
В инжектор хроматографа микрошприцем вводят 0,005 см3 экстракта пищевого продукта или БАД. На хроматограмме определяют площади пиков, соответствующие по временам удерживанию стандартным ДНС-производным биогенных аминов. Если пик на хроматограмме экстракта выходит за пределы шкалы, анализ проводят повторно после разбавления исходного раствора.
Вычисление результатов анализа
Содержание каждого биогенного амина в анализируемой пробе рассчитывают по формуле:
![]() ,
где
,
где
М - массовая концентрация БА, мг/кг или ppm,
т - массовая доля внешнего стандарта ДНС-амида, введенного для калибровки хроматографа, нг,
Sобр - площадь пика исследуемого компонента;
Scm - средняя площадь пика внешнего стандарта ДНС-амида, введенного для калибровки хроматографа;
![]() - общий объем
анализируемой пробы, мл;
- общий объем
анализируемой пробы, мл;
![]() - объем
экстракта, взятый для анализа, мл;
- объем
экстракта, взятый для анализа, мл;
![]() - объем, в
котором растворен сухой экстракт, мкл;
- объем, в
котором растворен сухой экстракт, мкл;
Va - объем экстракта, введенный в хроматограф, мл;
Р - навеска пробы, г.
Вычисления проводят до первого десятичного знака. За результат принимают среднее арифметическое результатов двух параллельных измерений и выражают целым числом с одним десятичным знаком.
Метрологические характеристики
Допустимое расхождение между результатами параллельных определений не должно превышать 10 % по отношению к среденеарифметическому значению.
Относительное стандартное отклонение при определении аминов составляет 0,06 - 0,1. Степень извлечения - 85,3 - 93,4 %.
Предел обнаружения метода 0,03 мг/кг для тирамина и 0,1 мг/кг для гистамина.
V. Метод определения полициклических ароматических углеводородов (ПАУ)
Сущность метода заключается в экстракции углеводородов из неомыляемой фракции липидов гексаном с последующей хроматографической очисткой, идентификацией и количественным определением ПАУ с помощью ВЭЖХ и спектрофлуориметрии, а также хромато-масс-спектрометрии.
Область применения метода распространяется в основном на БАД на основе рыбопродуктов, рыбьего жира и на основе растительных масел. Рекомендуемая масса пробы для анализа - 25 - 50 г.
Приведенные в методике варианты хроматографического разделения экстракта неомыляемой фракции липидов позволяют в зависимости от поставленной задачи реализовать имеющиеся в распоряжении аналитика приборы и реактивы.
В методику также включен как арбитражный - метод хромато-масс-спектрометрии.
Схема выделения полициклических ароматических углеводородов (ПАУ) включает переэкстракцию ПАУ из гексанового раствора неомыляемой фракции липидов комплексообразователем - диметилформамидом и хроматографическую очистку экстракта на колонке с сефадексом LH-20. Анализ ПАУ проводят с помощью ВЭЖХ или низкотемпературной спектрофлуориметрии. Ввиду того, что бенз(а)пирен рассматривают в качестве индикатора загрязнения ПАУ, в методику включены прописи его выделения и определения. Первый способ позволяет определить бенз(а)пирен в сумме ПАУ методом низкотемпературной спектрофлуориметрии, второй предполагает его выделение из суммы ПАУ в тонком слое ацетилированной целлюлозы и определение методом спектрофлуориметрии при комнатной температуре.
1. Специфические аппаратура, материалы и реактивы
Жидкостной хроматограф со спектрофотометрическим (фотометрическим) и флуориметрическим детекторами и колонкой с обращенно-фазовым сорбентом (силикагель ОDS или С18).
Хромато-масс-спектрометр с автоматической системой обработки данных, энергией ионизирующих электронов 70 eV, диапазоном массовых чисел 40 - 420 а.е.м., с капиллярной колонкой с неполярной или слабополярной жидкой фазой.
Спектрофотометр флуоресцентный с монохроматорами на линиях возбуждения и эмиссии.
Спектрофотометр флуоресцентный с приставкой для анализа флуоресценции и фосфоресценции при температуре жидкого азота (типа Hitachi-850, MPF-44B).
Спектрометр ДФС-12 или ДФС-24.
Облучатель с переменной длиной волны на 250 и 360 нм типа ОЛД ТУ 64-1-2242-77.
Пластины стеклянные 20´20 см для тонкослойной хроматографии.
Колонки стеклянные длиной 250 - 500 мм и внутренним диаметром 10 - 15 мм со стеклянным пористым фильтром ПОР 160 и шлифом 14.
Целлюлоза микрокристаллическая для хроматографии.
Силикагель для хроматографии 40 - 100 мкм.
Алюминия окись 5/40 мкм.
Сефадекс LH-20, «Фармация», Швеция.
Стандарты: флуорантен, пирен, бенз(а)антрацен, хризен, бенз(b)флуорантен, бенз(k)флуорантен, бенз(а)пирен, бенз(е)пирен, дибенз(а, h)антрацен, бенз(b)хризен, бенз(g, h, i)перилен, коронен, дибенз(а, е)пирен («Serva», Германия).
2. Подготовка к испытанию
2.1. Приготовление растворителей
Все используемые органические растворители перегоняют общепринятым способом.
Диметилформамид перегоняют, добавив к 250 см3 растворителя 12 см3 воды и 30 см3 бензола.
Отбирают фракцию, выкипающую выше 153 °С.
2.2. Подготовка сернокислого натрия безводного
1 кг сульфата натрия безводного заливают 2 дм3 перегнанного хлороформа или смеси хлороформа и метанола (1:1), оставляют на 3 - 4 ч или встряхивают в течение 1 ч на качалке в герметично закрытой колбе, суспензию отфильтровывают на воронке Бюхнера, промывая осадок на фильтре 1 дм3 растворителя, высушивают осадок на воздухе до полного удаления растворителя, после чего прокаливают при температуре 400 °С в течение 6 ч, хранят в колбе с притертой пробкой.
2.3. Приготовление адсорбентов для хроматографии
2.3.1. Приготовление ацетилированной целлюлозы для ТСХ
К 50 г микрокристаллической целлюлозы добавляют смесь 150 см3 бензола или толуола, 70 см3 уксусного ангидрида и 0,3 см3 концентрированной серной кислоты. Реакционную смесь перемешивают на магнитной мешалке в течение 6 - 8 ч, оставляют еще на 18 ч, после чего осадок отфильтровывают на воронке Бюхнера и заливают 300 см3 этанола, оставляют на 24 ч. Отфильтровывают осадок, промывают его на фильтре 100 см3 этанола, дистиллированной водой до нейтральной реакции (5´300 см3) и сушат на воздухе. Для приготовления пластинки 5 г ацетилированной целлюлозы суспендируют в 15 - 20 см3 этанола и наносят ровным слоем на стеклянную пластинку 20´20 см. После полного испарения растворителя проверяют хроматографическую подвижность бенз(а)пирена в системе этанол - ацетон - вода, взятых в объемном соотношении 60:25:15. Rf бенз(а)пирена и бенз(k)флуорантена составляет 0,1 и 0,2, соответственно.
2.3.2. 10 г сефадекса заливают 50 см3 этанола и оставляют для набухания на 3 - 4 ч, после чего переносят на колонку. Над сорбентом до начала работы должен быть слой растворителя 0,5 - 1,0 см.
2.4. Приготовление растворов сравнения, внутренних стандартов
Для приготовления исходных растворов берут навески не менее 5 мг, растворяют в бензоле в мерных колбах объемом 50 - 100 см3 и хранят в холодильнике. Рабочие растворы готовят из исходных разведением в соответствующем растворителе. Концентрации рабочих растворов внутренних стандартов и растворов сравнения следующие.
Внутренние стандарты для хромато-масс-спектрометрического анализа:
- растворы сравнения и внутреннего стандарта для анализа ПАУ (ВЭЖХ) - 100 мкг/см3.
Растворы сравнения для анализа ПАУ низкотемпературной спектрофлуориметрией:
- бенз(а)пирен - 1,0.., 10.., 100 нг/см3, перилен - 1.., 10 нг/см3, пирен, флуорантен, хризен, бенз(а)антрацен, бенз(b)флуорантен, бенз(k)флуорантен, дибенз(а, h)антрацен, дибенз(а, с)антрацен, бенз(е)пирен, бенз(g, h, i)перилен, дибенз(а, с)пирен, коронен - 1.., 10.., 100.., 1000 нг/см3 (в бензоле).
Концентрация раствора сравнения для определения бенз(а)пирена спектрофлуориметрией при комнатной температуре: бенз(а)пирен - 20... 100.., 1000 нг/см3.
3. Проведение испытания
3.1. Щелочной гидролиз и экстракция неомыляемой фракции липидов
50 г измельченного образца помещают в плоскодонную колбу вместимостью 1000 см3, добавляют раствор 19,5 г гидроокиси калия в 250 см3 92 %-ного этилового спирта, туда же помещают стеклянную трубочку-мешалку со впаянной стальной проволокой для перемешивания на магнитной мешалке. К реакционной смеси добавляют необходимые внутренние стандарты. Колбу присоединяют к обратному холодильнику и нагревают при кипении реакционной массы и перемешивании на магнитной мешалке в течение 3 ч. Полученный раствор охлаждают до комнатной температуры и через холодильник добавляют 350 см3 воды. Полученный раствор переносят в делительную воронку на 1000 см3 и экстрагируют трижды по 150 см3 гексана. Объединенные гексановые экстракты промывают в делительной воронке водой трижды по 50 см3, переносят в плоскодонную колбу, добавляют 30 - 40 г сульфата натрия и сушат экстракт в течение 24 ч. Полученный экстракт (экстракт А) - раствор неомыляемой фракции липидов, используют далее для выделения ПАУ.
3.2. Выделение ПАУ из неомыляемой фракции липидов (экстракта А)
3.2.1. Переэкстракция водным диметилформамидом
Экстракт А упаривают до объема 100 см3, переносят в делительную воронку емкостью 1000 см3 и добавляют 100 см3 смеси диметилформамида и воды, взятых в объемном соотношении 9:1. Содержимое воронки интенсивно встряхивают в течение 1 мин, отделяют нижний слой, а из верхнего гексанового экстрагируют ПАУ повторно, добавив в воронку новую порцию водного диметилформамида объемом 100 см3. Гексановый (верхний) слой отбрасывают, а объединенный диметилформамидный экстракт разбавляют 200 см3 воды и экстрагируют ПАУ из слоя разбавленного водой диметилформамида гексаном - трижды по 75 см3. Гексановые экстракты объединяют и промывают водой (4 раза по 50 см), отделяют и сушат с 30 г безводного сульфата натрия в течение 1 ч. Обезвоженный экстракт упаривают до объема 1 - 1,5 см3, остаток растворителя удаляют в токе азота. Вещество в колбе (экстракт В) растворяют в 1 - 1,5 см3 этанола и очищают на колонке с сефадексом LH-20.
3.2.2. Хроматография на колонке с сефадексом LH-20
10 г сефадекса заливают 50 см3 этанола и оставляют для набухания на 3 - 4 ч, после чего гель переносят на стеклянную хроматографическую колонку, дают растворителю стечь и переносят на колонку раствор экстракта В, смывая его из колбы 2 - 3 раза небольшим количеством этанола, каждый раз давая растворителю полностью впитаться в слой геля. Элюируют вещество с колонки этанолом со скоростью 0,5 см3/мин, обеспечивая такой поток небольшим избыточным давлением тока воздуха или азота, подаваемого на колонку через насадку со шлифом, заполненную силикагелем и слоем ваты. Первую фракцию - 50 см3 этанола отбрасывают, вторую фракцию - 100 см3 этанола, содержащую ПАУ с числом циклов 4 - 7, собирают, упаривают растворитель и далее анализируют с помощью ВЭЖХ, низкотемпературной спектрофлуориметрии или используют для выделения бенз(а)пирена.
3.2.3. Выделение бенз(а)пирена из фракции ПАУ ТСХ на ацетилированной целлюлозе
Фракцию ПАУ растворяют в 1 - 1,5 см3 бензола и наносят вещество сплошной полосой на пластинку с ацетилированной целлюлозой размером 20´20 см, подготовленную, как указано в п. 2.3.1, оставляя снизу и с боков пластинки поля шириной 1,5 - 2,0 см. На стартовую линию бокового поля, отделенного от основного с помощью скальпеля, наносят в точку 1 - 2 мкл раствора бенз(а)пирена, с концентрацией 1 мкг/см3. Пластинку помещают в хроматографическую камеру и проводят элюирование в смеси этанола - ацетон - вода (60:25:15). Камеру предварительно насыщают парами растворителя в течение 2 - 3 ч. Когда фронт растворителя достигнет 2 см от верхнего края пластинки, ее вынимают и отмечают зону бенз(а)пирена в УФ-свете с длиной волны 360 нм. Сорбент из зоны бенз(а)пирена (которая ниже зоны бенз(k)флуорантрена) собирают с помощью скальпеля на воронку Шотта и элюируют вещество бензолом (5´10 см3) в круглодонную колбу. Растворитель упаривают до небольшого объема, остаток отдувают в токе азота, затем добавляют 1 см3 бензола и анализируют с помощью спектрофлуориметрии при комнатной температуре.
3.3. Определение ПАУ с помощью ВЭЖХ
Рекомендуемые условия хроматографического разделения и детектирования ПАУ:
1) колонка Zorbax ODS 250´4 мм, подвижная фаза ацетонитрил - вода (84:16), скорость потока 1 см3/мин, детектор флуориметрический, длина волны возбуждающего света 280 нм, эмиссионного - 405 нм;
2) колонка та же, подвижная фаза метанол - вода (90:10), скорость потока 1,2 см3/мин, детектор спектрофотометрический при 254 или 290 нм;
3) колонка Supelcosil LC РАН 150´4,6 мм, подвижная фаза ацетонитрил - вода (80:20 или 85:15), скорость потока 1 см3/мин, детектор флуориметрический, длина волны возбуждающего света 300 нм, эмиссионного > 418 нм.
Объем анализируемой пробы определяют экспериментально. Он составляет не менее 200 мкл, объем вводимой пробы 10 - 20 мкл.
Расчет содержания компонентов суммы ПАУ (X) в образце проводят по следующим формулам:
а) по методу абсолютной калибровки:
![]() ,
где
,
где
Мст - количество компонента, введенного в хроматограф со стандартным раствором, мкг;
Scm - площадь пика компонента, введенного со стандартным раствором;
Sxo - площадь компонента на хроматограмме «холостого опыта»;
V1 - объем раствора пробы, мкл;
V2 - объем раствора пробы, введенной в хроматограф, мкл;
Sx - площадь пика компонента на хроматограмме пробы;
G - масса образца, г.
б) по методу внутреннего стандарта (добавка бенз(b)хризена)
![]() ,
где
,
где
Мст - количество введенного в образец внутреннего стандарта, мкг,
К - коэффициент пересчета, определяемый экспериментально в зависимости от выбранных условий детектирования по хроматограмме стандартной смеси ПАУ, включающей бенз(b)хризен. При длине волны возбуждающего света 360 нм и эмиссионного > 418 нм, этот коэффициент равен для:
- бенз(b)хризена - 1,00;
- пирена - 42,40;
- бенз(е)пирена - 26,10;
- бенз(b)флуорантена - 0,99;
- бенз(k)флуорантена - 0,30;
- бенз(а)пирена - 3,00;
- бенз(а)антрацена - 19,20.
3.4. Определение ПАУ методом низкотемпературной спектрофлуориметрии
3.4.1. Определение бенз(а)пирена в сумме ПАУ методом добавки
Фракцию ПАУ, полученную по п. 3.2.2, растворяют в 5 - 10 см3 бензола, из этого раствора в пробирку емкостью 10 см3 отбирают 1 см3 и добавляют к нему 2 см3 н-октана, получая таким образом раствор с нулевой добавкой бенз(а)пирена.
Спектры люминесценции получают на спектрометре дифракционном ДФС-12 или ДФС-24, а также на любом спектрофлуориметре с разрешением не менее 0,3 нм в области 380 - 450 нм.
При перекрытом потоке УФ-излучения закрепляют пробирку, опущенную в прозрачный сосуд Дьюара, перед щелью спектрометра; спустя 2 мин открывают доступ УФ-излучению к замерзшему раствору. Регулируя щели спектрометра, добиваются, чтобы интенсивность сигнала, создаваемого люминесценцией фона при длине волны 401,5 нм, не превышала 20 % шкалы. Записывают на спектрометре спектрограмму люминесценции в области 401,5 - 410 нм. Наличие в квазилинейчатом спектре люминесценции раствора фракции ПАУ полос с максимумом 403,0 нм (более интенсивная) и 408,5 нм (менее интенсивная) свидетельствует о присутствии бенз(а)пирена в этой фракции. Запись повторяют не менее двух раз при воспроизводимости не ниже, чем 10 % интенсивности сигнала при 403 нм. По интенсивности в максимуме люминесценции с длиной волны 403 нм в сравнении с интенсивностью этой линии в спектрах стандартных растворов бенз(а)пирена, записанных при тех же условиях возбуждения и регистрации, определяют концентрацию стандартного раствора бенз(а)пирена для анализа методом добавки. Если интенсивность линии 403 нм в спектре исследуемого раствора с нулевой добавкой несравнимо ниже, чем интенсивность этой линии в спектре стандартного раствора концентрации 1 нг/см3, исходный раствор фракции ПАУ концентрируют до меньшего объема и готовят новый раствор с нулевой добавкой, а затем повторно регистрируют его спектр и сравнивают высоту пика на спектрограмме с высотой пика той же линии в спектре стандарта. Если интенсивность линии 403 нм в спектре исследуемого раствора выше, чем в спектре стандартного раствора концентрации 100 нг/см3, то исследуемый раствор разбавляют в таком разведении, чтобы интенсивность линии 403 нм в получаемом растворе с нулевой добавкой не превышала интенсивности этой линии, даваемой указанным выше стандартным раствором.
Для определения концентрации исследуемого раствора далее готовят пробу с добавкой, соответствующей концентрации нулевого раствора. Для этого в пробирку на 10 см3 вносят 1 см3 исследуемого раствора в том же разведении, что и раствора с нулевой добавкой, а также 1 см3 н-октана и 1 см3 стандартного раствора с концентрацией, выбранной по результатам предварительной оценки, в диапазоне 0,1 - 100 нг/см3. Записывают спектрограмму исследуемого раствора с добавкой, как описано выше. При правильном выборе концентрации раствора-добавки высоты пиков полосы 403 нм исследуемого раствора и раствора с нулевой добавкой должны быть сравнимыми, при этом высота пика раствора с нулевой добавкой должна быть достоверно ниже. Если высота пика на спектрограмме раствора с добавкой на порядок выше, чем у раствора с нулевой добавкой, приготавливают новый раствор с добавкой меньшей концентрации. Если высоты пиков сравнимы для обоих растворов, в качестве добавки используется стандартный раствор большей концентрации (но не выше 100 нг/см3). Если высота пика на спектрограмме раствора с добавкой неизмеримо ниже высоты той же линии в спектре стандартного раствора той же концентрации, то это может быть связано с гашением люминесценции примесями. В этом случае раствор разбавляют н-октаном в 10 - 100 раз и повторяют запись спектров растворов с нулевой добавкой и с добавкой стандартного раствора соответствующей концентрации.
Измеряют на спектрограмме высоту спектральной линии в максимуме при 403 нм над уровнем фона, создаваемого люминесценцией примесей при 401,5 нм в растворе с нулевой добавкой (Y0 абсолютная) и высоту фона при той же длине волны над уровнем темнового тока (Y0 фона абсолютная) и вычисляют отношение:
![]() ,
где
,
где
![]() - относительная интенсивность аналитической линии в
растворе с 0-добавкой.
- относительная интенсивность аналитической линии в
растворе с 0-добавкой.
Аналогично вычисляют ![]() для повторной спектрограммы раствора с нулевой
добавкой и находят:
для повторной спектрограммы раствора с нулевой
добавкой и находят:
![]() .
.
Измеряют на спектрограмме высоту спектральной линии в максимуме при 403 нм над уровнем фона при 401,5 нм в растворе с концентрацией С0 (Y1 абсолютная) и высоту фона при 401,5 нм над уровнем темнового тока (Y1фона абсолютная) и вычисляют отношение этих величин:
![]() ,
где
,
где
Y1 - относительная интенсивность аналитической линии раствора с добавкой С концентрацией c0.
Аналогично вычисляют ![]() для повторной спектрограммы этого раствора и находят
среднее значение:
для повторной спектрограммы этого раствора и находят
среднее значение:
![]() .
.
Вычисляют концентрацию бенз(а)пирена в исследуемой фракции по формуле:
![]() (г/см3).
(г/см3).
Вычисляют содержание бенз(а)пирена (X) в образце по формуле:
![]() мкг/кг,
где
мкг/кг,
где
V1 - объем раствора фракции в бензоле, см3;
п - степень разбавления раствора (n = 1, если фракцию не разбавляют и не концентрируют для окончательного анализа);
0,8 - поправка на полноту извлечения;
G - масса образца, г.
4.7.2. Определение компонентов фракции ПАУ
Для количественного определения во фракции ПАУ 17 наиболее часто исследуемых соединений последовательно записывают спектры люминесценции раствора ПАУ при селективных для каждого из этих ПАУ длинах волн возбуждающего и эмиссионного света, приведенных в табл. 57.
Таблица 57
Спектры люминесценции ПАУ
|
lв |
lэм |
Соединение |
lв |
lэм |
|
|
Пирен |
338 |
372 |
бенз(k)флуорантен |
310 |
403 |
|
бенз(а)пирен |
298 |
403 |
дибенз(а, h)пирен |
313 |
448 |
|
Коронен |
307 |
444 |
бенз(е)пирен |
334 |
388 |
|
Фенантрен |
255 |
347 |
бенз(g, h, i)перилен |
303 |
420 |
|
Перилен |
414 |
444 |
флоурантен |
362 |
436 |
|
дибенз(а, h)антрацен |
301 |
394 |
бенз(b)флуорантен |
304 |
392 |
|
Хризен |
272 |
361 |
дибенз(а, с)антрацен |
291 |
375 |
|
Инденопирен |
362 |
463 |
дибенз(а, i)пирен |
398 |
431 |
|
бенз(а)антрацен |
292 |
384 |
|
|
|
При этом для каждого из 17 ПАУ готовят из исходного раствора фракции ПАУ серию из трех рабочих растворов, содержащих по 0,1 см3 исходного раствора, соответственно 0,1; 0,05 и 0,00 см3 н-октана и 0,00; 0,05 и 0,1 см3 эталонного раствора определяемого соединения с концентрацией, выбранной в зависимости от ориентировочно найденной его концентрации в исследуемом растворе. Для бенз(а)пирена диапазон концентраций эталонного раствора составляет 1 - 100 нг/см3, для перилена 1 - 10 нг/см3, для остальных ПАУ 1 - 1000 нг/см3. На полученных спектрограммах замеряют высоты спектральных линий в максимумах аналитических длин волн, приведенных в табл. 57, и определяют относительную интенсивность аналитических линий (аналогично п. 3.4.1). Расчет содержания компонентов фракции ПАУ в образце проводят по формуле, приведенной в разделе 3.4.1.
3.5. Определение бенз(а)пирена методом спектрофлуориметрии при комнатной температуре
Фракцию бенз(а)пирена, выделенную согласно п. 3.2.3, растворяют в 1 см3 бензола. В качестве раствора сравнения используют раствор, получаемый при добавлении 50 - 200 нг бенз(а)пирена к «холостому» опыту и разведенный в 1 см3 бензола. Записывают спектры флуоресценции этих растворов при длине волны возбуждающего света 388 нм в области 400 - 450 нм так, чтобы интенсивность в максимуме пика при 403 - 406 нм была не менее 25 % и не более 90 % шкалы. При необходимости изменяют разведение растворов и чувствительность прибора и снова записывают спектры в одном и том же режиме и при одинаковом разведении растворов.
Содержание бенз(а)пирена (X) в образце (мкг/кг) рассчитывают по следующей формуле:
![]() ,
где
,
где
С - концентрация раствора стандарта, мкг/см3;
Yx - интенсивность флуоресценции раствора пробы;
Ycm - интенсивность флуоресценции раствора сравнения;
V - объем пробы, см3;
G - масса образца, г;
0,75 - поправка на полноту извлечения.
4. Метрологические характеристики
Относительное допустимое расхождение между результатами двух параллельных определений, выполненных в одной лаборатории, по отношению к среднему арифметическому значению (Rr) и относительное допустимое расхождение между результатами испытаний, выполненных в двух разных лабораториях, по отношению к среднему арифметическому значению (RR), а также предел определения, по данным авторов методики, приведены в табл. 58.
Таблица 58
Относительные допустимые внутрилабораторные (Rr) и межлабораторные (RR) расхождения результатов определения и величина предела определения
|
Метод анализа |
Предел определения, мкг/кг |
Rr, % |
RR, % |
|
|
Полициклические ароматические углеводороды (индивидуальные соединения) |
ВЭЖХ с флуоресцентным детектором |
0,2 - 21 |
60 |
100 |
|
|
Низкотемпературный люминесцентный |
0,1 - 11 |
45 |
90 |
|
Бенз(а)пирен |
Спектрофлуориметрический |
0,1 |
45 |
90 |
VI. Показатели окислительной порчи масел
1. Перекисное число
Метод основан на реакции взаимодействия продуктов окисления растительных масел и животных жиров (перекисей и гидроперекисей) с йодистым калием в растворе уксусной кислоты и хлороформа с последующим количественным определением выделившегося йода раствором тиосульфата натрия титрометрическим методом по ГОСТ Р (ИСО 3960-1998).
Специфические приборы и реактивы
Весы лабораторные по ГОСТ 24104 2-го класса точности с наибольшим пределом взвешивания 200 г.
Колбы Кн-I-250-29/32 ТСХ по ГОСТ 25336.
Колба I (2)-1000-2 по ГОСТ 1770.
Стаканчики стеклянные цилиндрические для испытуемой пробы.
Бюретки 1-1(2, 3), 3-1(2)-5-0,02; 1-1(2, 3)-1(2)-10-0,05 по ГОСТ 29251.
Пипетки 2-2-1(2)-1 по ГСТ 29297.
Цилиндры 1(3)-25, 1(3)-100 по ГОСТ 1770.
Кислота уксусная по ГОСТ 61 х.ч., ледяная, не содержащая кислорода.
Хлороформ по ГОСТ 20015 свежеперегнанный, не содержащий кислорода.
Калий йодистый.
Натрий серноватистокислый (тиосульфат натрия).
Стандарт-титры тиосульфата натрия 0,1 г-моль.
Крахмал растворимый.
Вода дистиллированная.
Проведение испытания
Готовят раствор крахмала: 5 г растворимого крахмала смешивают с 30 см3; воды, добавляют 1000 см3 кипящей воды и кипятят в течение 3 мин.
Готовят раствор тиосульфата натрия из стандарт-титров (1 ампула на 1 дм3 дистиллированной воды). Для получения раствора тиосульфата натрия необходимых молярных концентраций 0,002 моль/дм3 и 0,01 моль/дм3 раствор разбавляют в 50 и 10 раз, соответственно. Разбавление проводят непосредственно перед использованием.
Раствор йодистого калия хранят в темном сосуде. Для приготовления 50 %-ного раствора KJ нужно 12,5 г поместить в колбу на 25 см3 и долить до метки дистиллированной водой.
В колбу со взвешенной пробой приливают 10 см3 хлороформа, быстро растворяют пробу, приливают 15 см3 уксусной кислоты и 1 см3 50 %-ного раствора йодистого калия, после чего колбу закрывают, перемешивают содержимое в течение 1 мин и оставляют на 20 мин в темном месте. Приливают в колбу 75 см3 воды, перемешивают и добавляют раствор крахмала до появления слабой однородной фиолетово-синей окраски. Выделившийся йод титруют раствором тиосульфата натрия до молочно-белой окраски, устойчивой в течение 5 сек.
Выполняют контрольное определение параллельно с основным определением.
Обработка результатов
Перекисное число вычисляют по формуле:
![]() ,
ммоль акт. кислорода/кг, где
,
ммоль акт. кислорода/кг, где
V - объем раствора тиосульфата натрия, использованный при определении, см3;
![]() - объем раствора
тиосульфата натрия, использованный при контрольном определении;
- объем раствора
тиосульфата натрия, использованный при контрольном определении;
С - действительная концентрация использованного раствора тиосульфата натрия, вычисленная с учетом поправки к номинальной концентрации;
т - масса испытуемой пробы, г.
За определение принимают среднее арифметическое значение результатов двух параллельных определений.
Расхождение между результатами двух независимых единичных определений, выполненных при использовании одного метода не должно превышать 10 % от среднего значения перекисного числа для перекисных чисел менее 3 ммоль акт. кислорода/кг.
2. Кислотное число
Метод основан на реакции нейтрализации свободных жирных кислот, содержащихся в маслах (ГОСТ 5476-64).
Специфические приборы и реактивы
Колбы конические вместимостью 250 мл по ГОСТ 10394-63.
Бюретки вместимостью 25 и 50 мл с делениями 0,1 мл по ГОСТ 1770-64.
Микробюретки вместимостью 2 мл с делениями 0,01 мм.
Весы технические.
Баня водяная.
Фенолфталеин, 1 %-ный спиртовой раствор.
Кали едкое, 0,1Н водный раствор.
Эфир этиловый.
Спирт этиловый технический.
Проведение испытания
В коническую колбу на технических весах отвешивают 3 - 5 г испытуемого масла, приливают 50 см3 нейтрализованной смеси (спирта с этиловым эфиром) и взбалтывают. Полученный раствор при взбалтывании быстро титруют 0,1Н раствором едкого кали до получения слабо-розовой окраски, устойчивой в течение 30 сек.
Обработка результатов
Кислотное число испытуемого масла (X) в мг вычисляют по формуле:
![]() ,
где
,
где
V - объем 0,1 н раствора едкого кали или едкого натра, израсходованного на титрование, в см3;
K - поправка к титру 0,1 н раствора едкого кали или едкого натра;
G - навеска испытуемого масла;
5,611 - постоянный множитель, независимо от применяемой щелочи.
Конечный результат выражается как среднее арифметическое между двумя параллельными определениями: при испытании масел не более 0,06 мг.
Приложение 1
Расчет метрологических характеристик методов и внутренний оперативный контроль качества результатов
Воспроизводимость (точность) - разброс относительно среднего результата, оценивают по отношению среднеквадратичного (стандартного) отклонения (sr) к среднему значению, выраженному в процентах. Правильность оценивают по измерению известного количества вещества в пробе и выражают в процентах. Сходимость - допустимое расхождение параллельных измерений при Р = 0,95, вычисляют по формуле 2,8×sr.
Относительная сходимость - отношение сходимости к среднему арифметическому значению параллельных измерений, выраженное в процентах.
Внутренний оперативный контроль качества результатов контрольного химического анализа (сходимость, воспроизводимость, точность) осуществляют с целью получения оперативной информации о качестве анализов и принятия при необходимости оперативных мер по его повышению (МИ 2335-95). Образцами для оперативного контроля воспроизводимости являются представительные пробы экстрактов (гидролизатов). Отбирают две пробы и каждую из них анализируют в точном соответствии с прописью методики, максимально варьируя условия проведения анализа, т.е. получают 2 результата анализа, используя разные наборы мерной посуды, разные партии реактивов.
Результаты контроля признаются удовлетворительными, если выполняется условие:
![]() ,
где
,
где
![]() - результат
анализа рабочей пробы, мкг/см3;
- результат
анализа рабочей пробы, мкг/см3;
![]() - результат
анализа этой же пробы, полученный другим аналитиком с использованием другой
мерной посуды и другой партии реактивов, мкг/см3;
- результат
анализа этой же пробы, полученный другим аналитиком с использованием другой
мерной посуды и другой партии реактивов, мкг/см3;
![]() - среднее
значение результатов двух анализов, мкг/см3;
- среднее
значение результатов двух анализов, мкг/см3;
D - допустимые расхождения между результатами анализа одной и той же пробы, соответствующие относительной сходимости каждого метода.
Образцами для оперативного контроля точности являются представительные пробы экстрактов (гидролизатов), к которым делаются добавки определяемого вещества в виде раствора. Отбирают две пробы и к одной их них делают добавку в виде раствора таким образом, чтобы его содержание увеличилось по сравнению с исходным на 50 - 150 %. Каждую пробу анализируют в точном соответствии с прописью методики и получают результат анализа исходной рабочей пробы X и рабочей пробы с добавкой X¢. Результаты анализа исходной рабочей пробы X и рабочей пробы с добавкой X¢ получают не по возможности, а в одинаковых условиях, т.е. их получает один аналитик с использованием одного набора мерной посуды, одной партии реактивов.
Результаты контроля признаются удовлетворительными, если выполняется условие:
![]() ,
где
,
где
С - добавка к пробе в виде раствора концентрацией, мкг/см3;
![]() - норматив
оперативного контроля погрешности, мкг/см3.
- норматив
оперативного контроля погрешности, мкг/см3.
При внешнем контроле (Р = 0,95) принимают
![]() ,
где
,
где
D2x¢ и D2х - характеристики погрешностей для исходной пробы и пробы с добавкой, мкг/см3.
Dx¢ = 0,165×X¢ и Dx = 0,165×X
При внутрилабораторном контроле (Р = 0,90) принимают
К¢д = 0,84×Кд
При превышении оперативного контроля погрешности эксперимент повторяют. При повторном превышении указанного норматива выясняют причины, приводящие к неудовлетворительным результатам контроля, и устраняют их.
СПИСОК ИСПОЛЬЗОВАННОЙ ЛИТЕРАТУРЫ
3. Carotenoids as colorants and vitamin A precursors. Technological and nutritional applications // Edited by J.C Bauernfeind. - New York: Academic Press, 1981 - P. 883 - 918.
4. Flavoring Substances and Natural Sources of Flavorings // 4th Edition Counsil of Europe. - Strasbourg. - 1992.
5. О. Isler, G. Brubacher. Vitamine. Part 1 // Georg Thieme Verlag, Stuttgart 1982. - 183 p.
6. Official Methods of Analysis of the АОAC, 15th ed. 1990. - Arlington.
7. Ollilalainen V., Finglas P.M., van den Berg H., Froidmontt-Gortz I. Certification of B-group vitamins in four food reference materials // J. Agr. And Food Chem. - 2001. - V. 49. - N 1. - P. 315 - 321.
8. «Виларин» ТУ 9361-002-00481152-99.
9. 865 душистых веществ для парфюмерии и бытовой химии / С.А. Войткевич. - М.: Пищевая промышленность. - 1994.
10. А. Лебедзинска, К.И. Эллер, В.А. Тутельян «Определение биогенных аминов в рыбе и рыбопродуктах с помощью нормально-фазовой высокоэффективной жидкостной хроматографии» // Журнал аналитической химии - 1989. - T. XLIV, вып. 5. - С. 928 - 931.
11. Кыргызский горный бальзам - мумие. / Б.К. Корчубеков, О.Н. Нарбеков. - Бишкек: Архар-Таш. - 1992.
12. ВФС 68-86-93. Таблетки мумие экстракта, покрытые оболочкой.
13. Голубкина Н.А., Прудник О.В., Журнал аналитической химии. - 1989. - T. XLIV, вып. 8. - С. 1349 - 1360.
14. ГОСТ 5476-64. Масла растительные. Метод определения кислотного числа.
15. ГОСТ Р (ИСО 3960-1998). Масла растительные. Метод определения перекисного числа.
16. ГОСТ Р 51483-99. Масла растительные и жиры животные. Определение методом газовой хроматографии массовой доли метиловых эфиров индивидуальных жирных кислот к их сумме.
17. ГОСТ Р 51486-99. Масла растительные и жиры животные. Получение метиловых эфиров жирных кислот.
18. ГОСТ 8558.1-78, ГОСТ 8558.2-78, ISO/DIS 6635, 1980. Мясные продукты. Методы определения нитрата и нитрита.
19. ГОСТ Р 50502-93. Напитки безалкогольные. Методы определения аспартама, сахарина, кофеина и бензоата натрия.
20. ГОСТ 24556-89. Продукты переработки плодов и овощей. Методы определения витамина С.
21. ГОСТ 29270-95. Продукты переработки плодов и овощей. Методы определения нитратов и нитритов.
22. ГОСТ Р 30711-2001. Продукты пищевые. Методы выявления и определения содержания афлатокчинов b1 и m1.
23. ГОСТ 26930-86. Сырье и продукты пищевые. Метод определения мышьяка.
24. ГОСТ 30178-96. Сырье и продукты пищевые. Атомно-абсорбционный метод определения токсичных элементов. Межгосударственный стандарт.
25. ГОСТ 26929-94, ГОСТ 26927-86. Сырье и продукты пищевые. Подготовка проб. Минерализация для определения содержания токсичных элементов. Межгосударственный стандарт.
26. ГОСТ 28038-89. Продукты переработки плодов и овощей. Метод определения микотоксина патулина.
27. ГОСТ 30627.2-98. Продукты молочные для детского питания. Методы измерений массовой доли витамина С (аскорбиновой кислоты).
28. ГОСТ Р 51650-2000. Продукты пищевые. Методы определения массовой доли бенз(а)пирена.
29. ГОСТ 9517-94. Топливо твердое. Методы определения выхода гуминовых кислот.
30. Госфармакопея СССР. - XI изд., т. 1 - 2.
31. Дополнение № 4274-87 к СанПиН 42123-4083-86. Временные гигиенические нормативы и метод определения содержания гистамина в рыбопродуктах.
32. Европейская фармакопея, 1997: 0957.
33. Ляпков Б.Г., Воробьева Л.Ш., Медведев Ф.А. и др. / Экстракционное концентрирование и хромато-масс-спектрометрическое определение D-лимонена в маслах цитрусовых и других биологических образцах // ЖАХ. - 1996, т. 51, № 4, С. 451 - 454.
34. Методы исследования эфирных масел / Н. Гаряев, И. Плива. - Алма-Ата. - 1962.
35. МУ 4721-88. Методические указания по выделению, идентификации и количественному определению насыщенных, моно-, би-, три- и ряда полициклических ароматических углеводородов в пищевых продуктах.
36. МУ 5178-90 «Методические указания по определению ртути в пищевых продуктах методом беспламенной атомной абсорбции».
37. МУК 4.4.1.011-93. Методы определения летучих N-нитрозаминов в пищевых продуктах и продовольственном сырье.
38. Немецкая фармакопея 1997 (DAB 10).
39. Руководство по методам анализа качества и безопасности пищевых продуктов / Под. ред. И.М. Скурихина, В.А. Тутельяна. - М.: Брандес-Медицина, 1998.
40. Справочник биохимика: Пер. с англ./ Досон Р., Эллиот Д., Эллиот У., Джонс К.-М.: Мир, 1991. - 544 с.
41. Теоретические и клинические аспекты науки о питании. Т. 8. Методы оценки обеспеченности населения витаминами. // Под ред. Волгарева М.Н. - М. 1987. - С. 173 - 184.
42. Французская фармакопея, X изд., т. 2, 1990.
43. ФС 42-2725-90.
44. Эллер К.И., Пименова В.В., Левин Л.Г., Киселева М.Г. Комплексный подход к оценке подлинности окрашенных соков // Вопр. питания. - 2003, № 2, с. 28 - 35.
45. Якушина Л.М., Бекетова Н.А., Бендер Е.Д., Харитончик Л.А. Использование методов ВЭЖХ для определения витаминов в биологических жидкостях и пищевых продуктах. // Вопр. питания. - 1993, № 1. - С. 43 - 47.